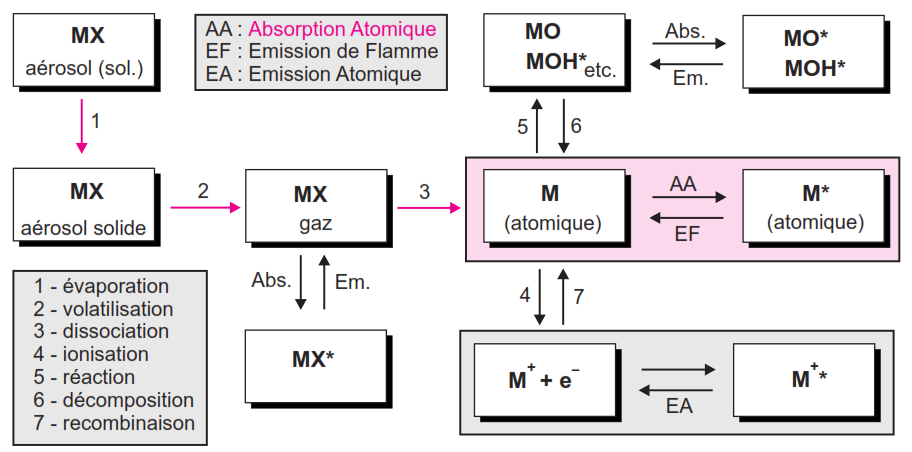
La spectrophotométrie d’absorption atomique
Mesures
Préparation de l’échantillon
On travaille toujours en solution. Si l’échantillon est solide (mise en solution ou en suspension ou minéralisation).
Ce sont des solutions aqueuses avec, parfois, addition d’un solvant organique. Ces solutions sont souvent très diluées.
Dans le cas d’une matrice solide, on passe le plus souvent par une mise en solution. Pour cette étape on fera appel , suivant l’analyte recherché, soit à une minéralisation, soit à une extraction (cours méthodes d’extraction).
Les méthodes de minéralisation sont des méthodes agressives qui ont pour but de détruire les composés organiques et les constituants biologiques de la matrice. Les éléments sont libérés sous une forme ionique ou moléculaire simple.
On distingue deux grandes voies de minéralisation: la minéralisation par voie humide, également nommée digestion acide, et la minéralisation par voie sèche ou calcination.
- Minéralisation par voie humide : On utilise un volume de réactif (des acides, tels que H2SO4, HNO3, H3PO4, HCl, l’eau régale (mélange de HNO3 et HCl dans un rapport de 1:3), HF et/ou des oxydants, tels que H2O2 , HClO4, H2SO4, HNO3) en excès par rapport à la prise d’essai, en chauffant à une température relativement basse(de 100 à 200
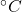 ) durant un temps assez court (quelques heures au plus).
) durant un temps assez court (quelques heures au plus). - Minéralisation par voie sèche : On chauffe à des températures bien supérieures (vers 450 à 550, parfois plus), pendant un temps beaucoup plus long, de l’ordre de la journée ou plus, en utilisant une faible quantité de réactif, généralement un oxydant (H2SO4, HNO3, K2SO4, KNO3, Mg(NO3)2, MgO …) pris comme aide de minéralisation.
Chacune de ces voies de minéralisation a ses avantages et ses inconvénients. Le choix d’une méthode de minéralisation est étroitement lié à celui de la méthode de mesure mais il dépend aussi de la matrice, de l’analyte et de sa forme physicochimique.
Dosage
La spectrométrie d’absorption atomique est une méthode analytique comparative ; elle implique un étalonnage.
Étalonnage externe
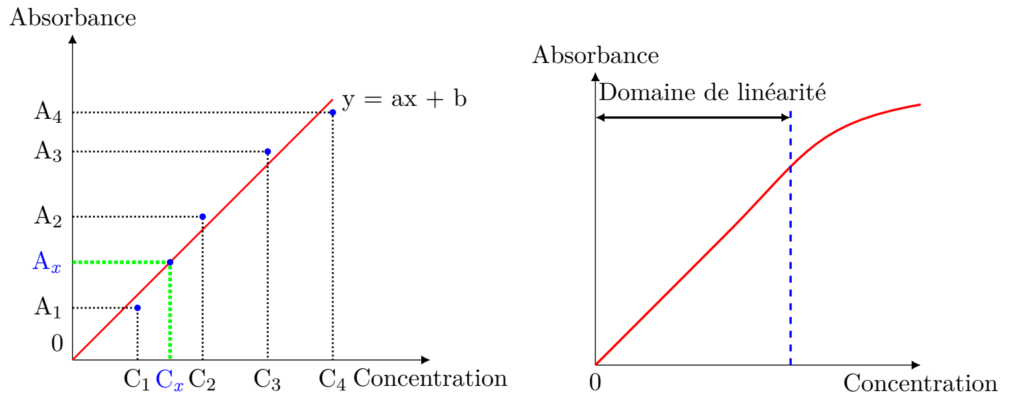
L’étalonnage le plus courant s’obtient en mesurant l’absorbance de solutions à concentrations progressives et connues en analyte.
La concentration de la solution inconnue est alors directement déduite en rapportant sa valeur d’absorbance sur une droite d’étalonnage préalablement établie. Cette méthode, appelée étalonnage direct ou étalonnage externe, s’applique à des milieux relativement simples, dont la matrice est suffisamment constante. Il faut toujours vérifier que le domaine des mesures est linéaire .
On prépare ainsi un certain nombre de solutions de concentrations connues : 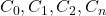 , et la solution de concentration inconnue
, et la solution de concentration inconnue 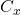 . On mesure ensuite les absorbances
. On mesure ensuite les absorbances 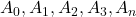 et on détermine la concentration à partir de l’absorbance
et on détermine la concentration à partir de l’absorbance 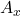 de l’échantillon.
de l’échantillon.
On recherche la fonction : y = ax + b. Avec y : Absorbance, x: la concentration de l’élément métallique.
Étalonnage par ajouts dosés
Si la matrice est inconnue ou trop variable dans la série d’échantillons analysés, on dispose de la méthode des ajouts dosés. Elle consiste à additionner des quantités croissantes et connues de l’élément considéré Qi à la solution étudiée. Le volume de ces ajouts successifs doit être suffisamment faible pour que l’on puisse négliger les variations de volume de la solution d’échantillon, On obtient des concentrations croissantes Ci’.
On mesure ensuite l’absorbance des solutions obtenues. En traçant la courbe Absorbance= f(concentration), on obtient une droite dont l’intersection avec l’axe des abscisses donne la concentration de l’analyte dans la solution sans ajout. Actuellement, les appareils modernes équipés d’un distributeur d’échantillons peuvent effectuer ces opérations de façon entièrement automatique.
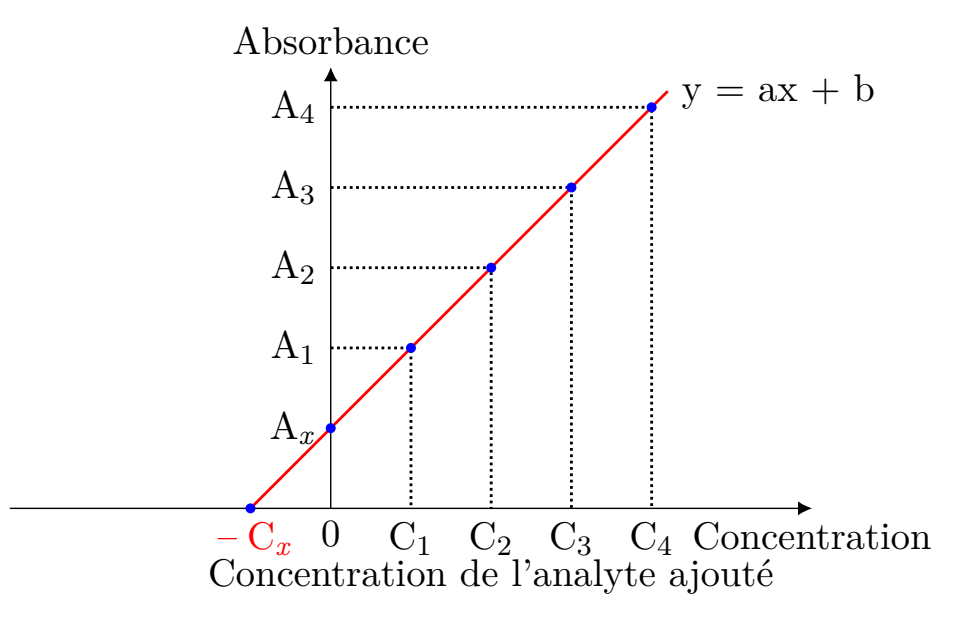
Il faut autant de gammes que d’échantillons, mais l’avantage est que cette méthode tient compte de l’effet de matrice :
un certain nombre de substances sont susceptibles de donner des interférences qui, ici, portent à la fois sur l’échantillon et les étalons.
Conséquences : on élimine l’erreur due aux interférences.
Interférences
On appelle perturbation, interaction ou interférence, l’influence d’un ou de plusieurs constituants du milieu analysé sur le dosage d’un élément.
La concentration de l’analyte déterminée sans tenir compte d’une interaction est appelée concentration apparente ; celle-ci peut être plus élevée que la concentration réelle, il y a alors exaltation ; dans le cas contraire, dépression. Les perturbations en SAA sont classées de trois manières :
- Interférences spectrales
- Interférences chimiques
- Interférences physiques
Interférences spectrales
Appelées aussi absorptions non spécifiques, elles sont dues aux phénomènes ayant leur siège dans la source d’atomisation et affectant la mesure spectrale d’absorbance de l’analyte, par suite d’une superposition de raies, de la présence de bandes d’absorption moléculaire ou d’une diffusion de la lumière incidente sur des particules solides ou liquides présentes dans l’atomiseur.
Le détecteur, quel qu’il soit, ne peut mesurer qu’une intensité lumineuse. Il faut donc convertir ce signal en absorbance et, en plus, corriger cette absorbance des absorptions non spécifiques (ANS).
Cela implique que le détecteur doit enregistrer un grand nombre de signaux en un temps très court et que l’électronique située en aval du détecteur doit traiter ces signaux très rapidement.
Les différents signaux à traiter sont les suivants :
- Intensité incidente I0
- Intensité émise par l’atomiseur E
- Intensité émergente I + E
À ces trois mesures de base, indispensables à la détermination de l’absorbance, s’ajoutent celles permettant la correction des absorptions non spécifiques (ANS) :
- Intensité incidente du correcteur (elle vaut aussi I0)
- Intensité émergente après les absorptions non spécifiques I‘
Au départ des trois premières mesures, l’électronique va convertir ces signaux en absorbance. Il s’agira de l’absorbance totale (spécifique et non spécifique). Les mesures de I‘ combinées à celles de E et de I0 seront converties en absorbance non spécifique. Cette ANS est ensuite soustraite de l’absorbance totale afin d’obtenir finalement l’absorbance spécifique.
Diffusion de la lumière incidente
Dans de très nombreux cas, l’atomisation de l’analyte en spectroscopie d’absorption atomique électrothermique (SAAE) s’accompagne de fumées constituées de particules solides ou liquides provenant de la décomposition de l’échantillon ou de la recombinaison des atomes et des molécules de la phase gazeuse. De telles particules, n’étant pas évacuées par le gaz de balayage qui est interrompu lors de cette étape, ont pour effet de diffuser dans toutes les directions la lumière issue de la source primaire.
On observe alors une absorption continue dans tout le domaine spectral, se superposant à l’absorption atomique de l’analyte. Ainsi, une matrice chargée en NaCl donnera naissance, au moment de la recondensation de la vapeur de l’halogénure, à des particules (NaCl) solides ou liquides qui diffusent la lumière, si elles sont encore présentes dans le tube lors de la mesure de l’absorption atomique. Les absorptions par diffusion de lumière sont, pour des niveaux raisonnables, efficacement compensées par tous les systèmes de correction de fond (deutérium, Zeeman, Smith – Hieftje).
En spectroscopie d’absorption atomique de flamme (SAAF), des problèmes similaires peuvent apparaître suite à la présence dans la flamme de gouttelettes non vaporisées ou de microcristaux non dissociés.
Élargissement de la raie d’émission de la source (effets Doppler-Fizeau, Lorentz, Stark)
Se traduit par les effets de la différence de potentiel (ddp) entre les électrodes : (T°C), champ électrique. Si la ddp est trop importante, on a élargissement de la raie qui peut être plus large que la raie d’absorption de l’élément à doser.
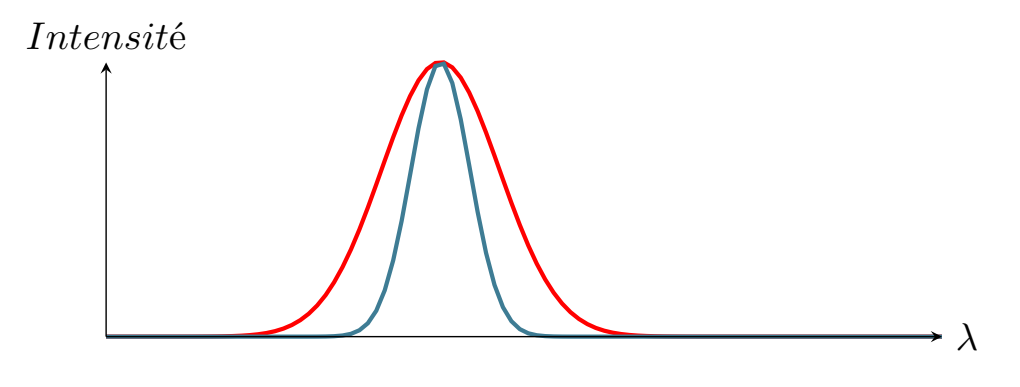
Absorption non spécifique
Les molécules dans la flamme sont susceptibles d’absorber le faisceau incident, On a ainsi une erreur par excès.
Certaines molécules provenant de la matrice de l’échantillon analysé présentent un spectre d’absorption moléculaire comprenant de larges bandes continues, spécifiques de l’espèce chimique, et localisées entre 200 et 350 nm.
Les systèmes de correction disponibles sur le marché apportent une solution satisfaisante pour tous les cas courants.
Correction des absorptions non spécifiques
Le rôle des correcteurs est de mesurer automatiquement les ANS dues aux interférents en tout genre afin de les soustraire de l’absorbance totale. Pour que la correction soit valable, il faut qu’elle soit effectuée aussi près que possible de la longueur d’onde caractéristique 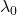 .
.
La correction par lampe au deutérium
La source au deutérium D2 est une source continue. Il y a une alternance des 2 sources (source primaire de la lampe à cathode creuse (LLC) et la source au D2).
Limité à l’ultraviolet 
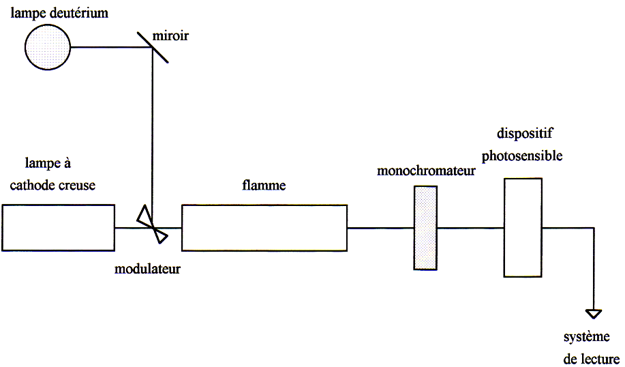
Lorsque l’appareil est équipé d’un correcteur au deutérium, une lampe au deutérium, produisant une lumière continue dans tout l’ultraviolet, est placé de telle sorte que les chemins optiques des lumières provenant des deux sources soient identiques. L’atomiseur (flamme ou four) est éclairé alternativement par les deux rayonnements. Nous pouvons schématiser le fonctionnement du correcteur au deutérium de la manière suivante.
- La source primaire SP (cathode creuse ou autre) émet un spectre de raies tandis que la source Lorsque l’appareil est équipé d’un correcteur au deutérium, une lampe au deutérium, produisant une lumière continue dans tout l’ultraviolet, est placé de telle sorte que les chemins optiques des lumières provenant des deux sources soient identiques. L’atomiseur (flamme ou four) est éclairé alternativement par les deux rayonnements. Nous pouvons schématiser le fonctionnement du correcteur au deutérium de la manière suivante, source continue SC, émet un spectre continu (figure a).
- Le monochromateur sélectionne la raie caractéristique de l’analyte (figure a SP) tandis qu’il laisse passer la lumière de la source continue sur toute la largeur de la bande passante (figure a SC). On peut admettre que l’intensité lumineuse en chaque point de la bande passante est la même.
- Les deux intensités intégrées incidentes (
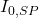 et
et 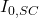 ) sont égalées. L’une, , est représentée par la largeur de la raie (
) sont égalées. L’une, , est représentée par la largeur de la raie ( 5
5 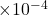 nm), l’autre, , est répartie sur la bande passante (1 à 2 nm) (figure a) =
nm), l’autre, , est répartie sur la bande passante (1 à 2 nm) (figure a) = - Lorsque c’est la source primaire qui éclaire l’atomiseur, la lumière est atténuée par les absorptions spécifiques (figure b SP) et non spécifiques (figure c SP). Soit
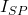 l’intensité transmise de la source primaire,
l’intensité transmise de la source primaire, 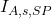 , l’intensité absorbée par les absorptions spécifiques et
, l’intensité absorbée par les absorptions spécifiques et 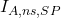 , celle absorbée par les absorptions non spécifiques
, celle absorbée par les absorptions non spécifiques 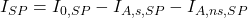
- Lorsque c’est la lampe au deutérium qui éclaire l’atomiseur, les mêmes absorptions se produisent (figure b SC et c SC) :
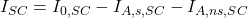 , mais comme la raie atomique est très étroite (figure b SC)
, mais comme la raie atomique est très étroite (figure b SC) 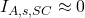 , d’où :
, d’où : 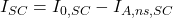 .
. - Si le fond est réellement continu, les ANS ont lieu dans les mêmes proportions que lors de l’éclairage par la source primaire (figure c) :
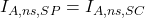 .
. - L’électronique en aval du détecteur a donc en mémoire
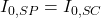 , et
, et 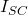 .
.
(1) 
L’utilisation du correcteur au deutérium implique la superposition des deux faisceaux incidents.
Le correcteur au deutérium ne peut corriger les ANS qu’entre 190 et 390 nm et la correction n’est valable que si les ANS sont inférieures à un certain niveau.
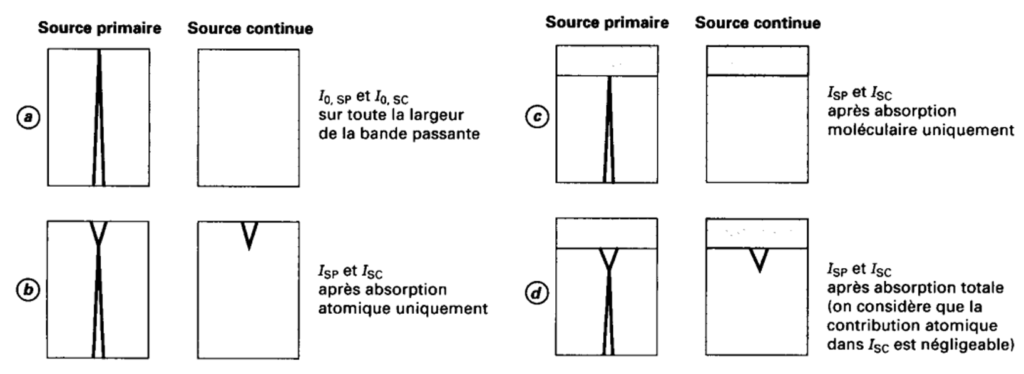
La correction par effet Smith-Hieftje
Le fonctionnement du correcteur est basé sur une alimentation particulière de la lampe à cathode creuse en deux phases. Pendant la première phase, la lampe est alimentée normalement (courant faible) et la raie d’émission est normale ; ensuite, durant un court instant, elle est suralimentée (courant élevé). Dans ce cas, la densité du nuage atomique à l’extrémité de la cathode augmente et les atomes peuvent absorber les photons qui sortent de la cathode. On observe également un élargissement de la raie d’émission suite à l’élévation du courant.
Le centre de la raie d’émission est ainsi absorbé et celle-ci apparaît alors comme dédoublée. Ce phénomène est connu sous le nom de renversement de raie.
Voyons maintenant ce qui se passe au niveau de l’atomiseur.
Lorsque la cathode creuse est alimentée normalement, on mesure l’absorbance totale :
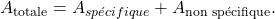
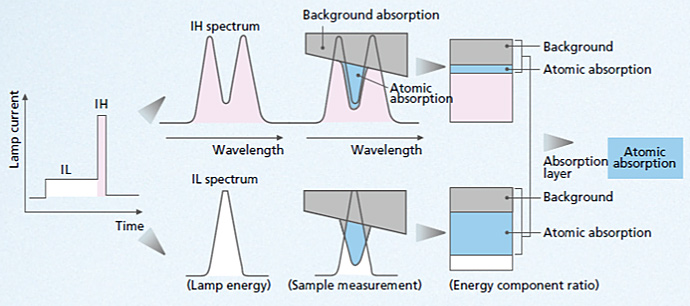
Lorsque la lampe est suralimentée, si le renversement de raie est suffisant, les deux composantes seront réparties également de part et d’autre de la raie d’absorption atomique. On mesure dès lors uniquement le fond. Par soustraction, on obtient l’absorbance spécifique.
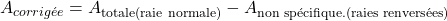
Du fait qu’il n’y a qu’une seule source de lumière et que le doublet est symétrique, les problèmes d’alignement et de réglage des intensités incidentes ont disparu.
La correction par effet Zeeman
L’effet Zeeman est un phénomène qui concerne les niveaux énergétiques d’un atome et, par voie de conséquence, les transitions électroniques, donc, mais indirectement, les photons émis ou absorbés.
Effet Zeeman normal
Prenons le cas simple où l’état fondamental est un état S et l’état excité, un état P. En l’absence de champ magnétique (figure a), les deux niveaux de l’atome sont uniques et la seule transition possible est le passage de 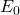 à
à 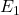 (absorption) ou de à (émission), entraînant l’absorption ou l’émission d’un photon unique de fréquence
(absorption) ou de à (émission), entraînant l’absorption ou l’émission d’un photon unique de fréquence 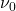 et de longueur d’onde . Le niveau excité P est en fait dégénéré de telle sorte que si l’atome est placé dans un champ magnétique, ce niveau va se diviser en trois sous-niveaux, suivant la valeur du nombre quantique magnétique. Un de ces niveaux, , est exactement à la même énergie qu’en l’absence de champ magnétique. Les deux autres sont répartis symétriquement de part et d’autre du niveau central. Ce sont les niveaux
et de longueur d’onde . Le niveau excité P est en fait dégénéré de telle sorte que si l’atome est placé dans un champ magnétique, ce niveau va se diviser en trois sous-niveaux, suivant la valeur du nombre quantique magnétique. Un de ces niveaux, , est exactement à la même énergie qu’en l’absence de champ magnétique. Les deux autres sont répartis symétriquement de part et d’autre du niveau central. Ce sont les niveaux 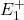 et
et 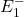 .
.
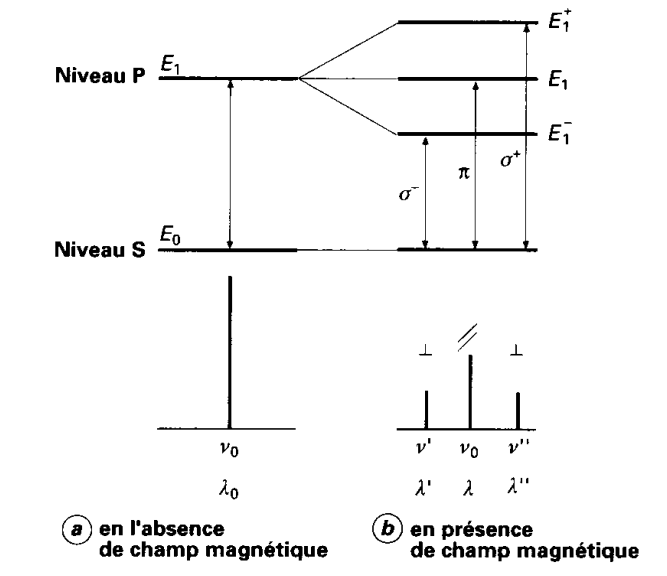
L’état fondamental reste unique. Il en résulte que, en présence d’un champ magnétique, trois transitions sont possibles, en absorption comme en émission. L’une de fréquence ( → ) et de longueur d’onde 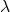 (
(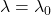 ) est identique à celle observée en l’absence de champ magnétique. Une autre est à la fréquence
) est identique à celle observée en l’absence de champ magnétique. Une autre est à la fréquence 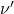 , soit à la longueur d’onde
, soit à la longueur d’onde 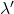 . Enfin la dernière est à la fréquence
. Enfin la dernière est à la fréquence 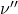 et à la longueur d’onde
et à la longueur d’onde 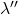 . L’effet Zeeman normal n’est observé que pour quelques éléments, les alcalino-terreux et les éléments du groupe du zinc (groupe II a et II b). Dans d’autres cas, il y a un détriplement de la raie spectrale alors que la démultiplication des niveaux est beaucoup plus élevée. C’est ainsi qu’aux éléments déjà cités s’ajoutent Pb, Si, V, Pd, Sn. Pour aborder l’aspect théorique de l’effet Zeeman normal, introduisons quelques définitions :
. L’effet Zeeman normal n’est observé que pour quelques éléments, les alcalino-terreux et les éléments du groupe du zinc (groupe II a et II b). Dans d’autres cas, il y a un détriplement de la raie spectrale alors que la démultiplication des niveaux est beaucoup plus élevée. C’est ainsi qu’aux éléments déjà cités s’ajoutent Pb, Si, V, Pd, Sn. Pour aborder l’aspect théorique de l’effet Zeeman normal, introduisons quelques définitions :
- Montage direct : la source primaire est soumise au champ magnétique et la raie d’émission est démultipliée.
- Montage inverse : l’atomiseur est soumis au champ magnétique et la raie d’absorption est démultipliée.
- Montage longitudinal : la direction d’observation est parallèle au champ magnétique.
- Montage transverse : la direction d’observation est perpendiculaire au champ magnétique.
- Le champ peut être constant (aimant permanent ou électroaimant à courant continu), ou alternatif (électroaimant à courant alternatif).
Les trois raies 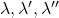 , possèdent un certain nombre de propriétés.
, possèdent un certain nombre de propriétés.
- La somme des intensités de ces trois raies est égale à l’intensité de la raie unique en l’absence de champ magnétique.
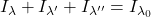
- Les intensités relatives de ces trois raies sont respectivement dans le cas du montage transverse :
(2)
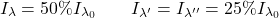
Par contre, dans le montage longitudinal, la raie est absente : (3)
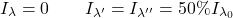
- La lumière émise à ces trois longueurs d’onde est polarisée. La polarisation dépend des directions relatives du faisceau lumineux et du champ magnétique. Si le montage est transverse, la raie [/latex]\lambda[/latex] est polarisée parallèlement au champ magnétique et est appelée raie
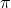 , tandis que, aux autres longueurs d’onde, la lumière est polarisée perpendiculairement à la direction du champ magnétique ; les raies sont alors appelées respectivement
, tandis que, aux autres longueurs d’onde, la lumière est polarisée perpendiculairement à la direction du champ magnétique ; les raies sont alors appelées respectivement 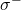 et
et  . Si le montage est longitudinal, il ne reste que les deux composantes et qui sont polarisées circulairement, l’une à gauche, l’autre à droite. De la même manière, les raies d’absorption sont polarisées.
. Si le montage est longitudinal, il ne reste que les deux composantes et qui sont polarisées circulairement, l’une à gauche, l’autre à droite. De la même manière, les raies d’absorption sont polarisées. - L’écartement entre les trois raies est d’autant plus important que le champ magnétique est intense. Il faut que le champ magnétique soit supérieur à 0,8 Tesla pour qu’il n’y ait plus de chevauchement entre les raies et
 .
.
Effet Zeeman anormal
Pour ce qui est des autres éléments qui ne présentent pas un état singulet S, la démultiplication des raies est beaucoup plus importante : c’est l’effet Zeeman anormal. Il reste des composantes , des composantes et des composantes . Suivant les cas, il y aura 4, 5 ou 6 composantes dans chacun des groupes et . Les propriétés des ensembles et sont les mêmes que dans l’effet normal.
Par contre, en ce qui concerne la présence d’une raie à la longueur d’onde , il y a des différences suivant les éléments.
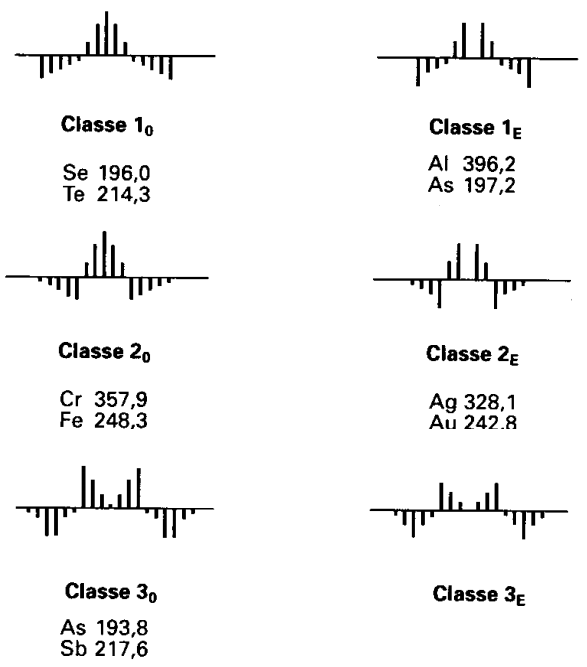
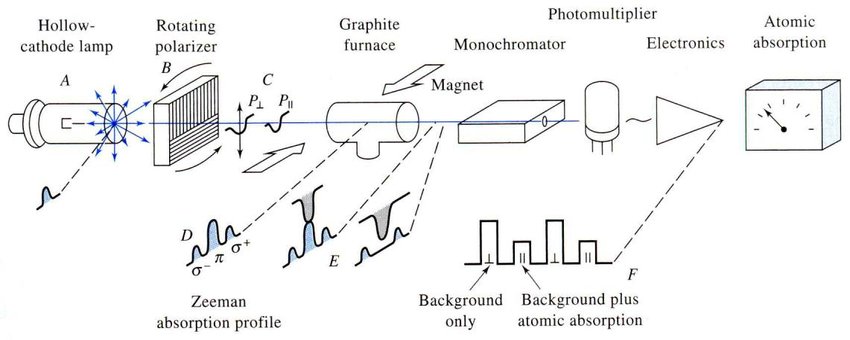
Le principe de la correction pour l’effet Zeeman normal sur un montage direct – transverse – constant est comme suit:
La lampe à cathode creuse est placée dans un champ constant et transverse. Les niveaux énergétiques des atomes produits dans la lampe à cathode creuse sont démultipliés de telle sorte que l’émission est décomposée en trois raies, une (), et deux qui sont légèrement décalées. Un quartz piézoélectrique tournant est placé entre la source et l’atomiseur. L’atomiseur est donc éclairé alternativement par la raie et par les raies . La raie d’absorption atomique, dans l’atomiseur, n’a subi aucune modification et sa longueur d’onde correspond à celle de la raie ().
Lorsque le polariseur est parallèle au champ, la raie passe dans l’atomiseur. La raie est atténuée par l’élément à analyser (absorption spécifique) et aussi les autres composés de la matrice (absorption non spécifique). Si le champ magnétique est suffisamment intense, les raies ne chevauchent pas la raie d’absorption atomique. Lorsque le polariseur est perpendiculaire, les raies éclairent l’atomiseur et sont atténuées uniquement par le fond continu. Il y a d’autres montages autres que le montage décrit ici.
Cette technique de correction est:
- La plus répandue
- Décomposition des niveaux énergétiques par un champ magnétique.
-
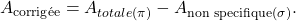
- Opère à toutes les longueurs d’onde (193 – 852 nm)
Interférences chimiques
Elles sont dues à la formation dans la flamme d’oxydes réfractaires ou de sels peu volatils. On parle aussi d’effet de matrice. Elles résultent des modifications, dans la source d’atomisation, des processus de dissociation, d’oxydoréduction ou d’ionisation. Cela est dû à l’altération du nombre ou de la vitesse de formation d’atomes de l’analyte, altération qui résulte de la perturbation dans le lieu d’atomisation.
Elles sont dues à la formation d’oxydes réfractaires ou de sels peu volatils dans la flamme.
Formation d’oxydes
(4) 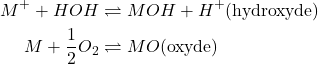
Exemple: 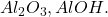
Pour éviter ce phénomène on utilise une flamme dite réductrice:
- faible teneur en oxygène dans le gaz.
- utiliser le protoxyde d’azote
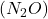 comme oxydant
comme oxydant
Formation de sel réfractaire
(5) 
Pour éliminer ces sels réfractaires tel que Ca_3(PO_4)_3} on ajoute un sel (tampon spectral) dont le cation est capable de former des sels encore plus insolubles et susceptibles de donner des sels plus volatils avec l’élément à doser.
Les sels les plus utilisés sont:
- Chlorure de strontium:
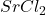
- Chlorure de lanthane:
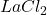
Interférences physiques
Elles sont généralement liées aux propriétés physiques des solutions étudiées. Ce type d’interférences se remarque plutôt en SAAF où l’introduction de l’échantillon est assurée par un système de nébulisation ; un changement, par exemple, de la viscosité entre les étalons et les échantillons peut apporter des erreurs. En SAAE, les interférences de transport ne se limiteront qu’aux erreurs entraînées par les différences de viscosité entre les étalons et les échantillons, mais dans une moindre mesure qu’en SAAF.
Pour corriger ces interférences on:
- Rend la solution iso-visqueuse: addition d’albumine, ou d’agents mouillants.
- Utilise la méthode d’étalonnage à ajouts dosés (ajouts connus).
Automatisation
En SAA, l’automatisation intervient essentiellement au niveau de l’introduction de l’échantillon dans l’atomiseur. Les passeurs automatiques d’échantillons sont capables de prélever les solutions mais également d’effectuer les rinçages intermédiaires, les dilutions éventuelles, les additions de solutions étalons (ajouts dosés).
Applications
La spectrométrie d’absorption atomique peut être appliquée au dosage d’une trentaine d’éléments, et cela tout aussi bien au niveau des éléments majeurs (domaine analytique de la SAAF : 0,1 à 10 mg/L) que d’éléments traces ou ultratraces (domaine analytique de la SAAE : 0,0001 à 0,1 mg/L). La SAA couvre un vaste éventail d’applications : l’analyse des eaux, des tissus végétaux et animaux, des aliments et boissons, des sols, engrais et sédiments, des liquides biologiques, médicaments, des produits industriels (ciments, verres, métaux, produits pétroliers…).
Biologie
Dans le domaine de l’analyse médicale, le contrôle des métaux dans les liquides biologiques est très important. Les deux types d’échantillons les plus analysés sont le sang et l’urine.
- Dosage du: Ca2+ , Mg2+
- Dosage des oligo-éléments: Zn2+, Cu2+ , Ce2+
Toxicologie
- Dosage des métaux lourds : Al, Cr, Cd, Hg dans diverses matrices.
- Dosage du Pb dans le sang (suspicion de saturnisme)
Pharmacie
La SAA est inscrite à la pharmacopée européenne comme technique d’analyse, elle est recommandée par exemple dans la détermination de la teneur :
- de l’Aluminium dans les solutions de dialyse péritonéale. Il faut vérifier la teneur en aluminium (risque d’encéphalopathie).
- du Zinc dans les préparations injectables d’insuline.
- du Cadmium, Plomb et Nickel dans le stéarate de magnésium
Environnement
- Dosage des métaux toxiques dans l’eau, plantes et poissons …
- Dosage des métaux toxiques dans le sol, roches, sédiments …
Share this content:
Table des matières