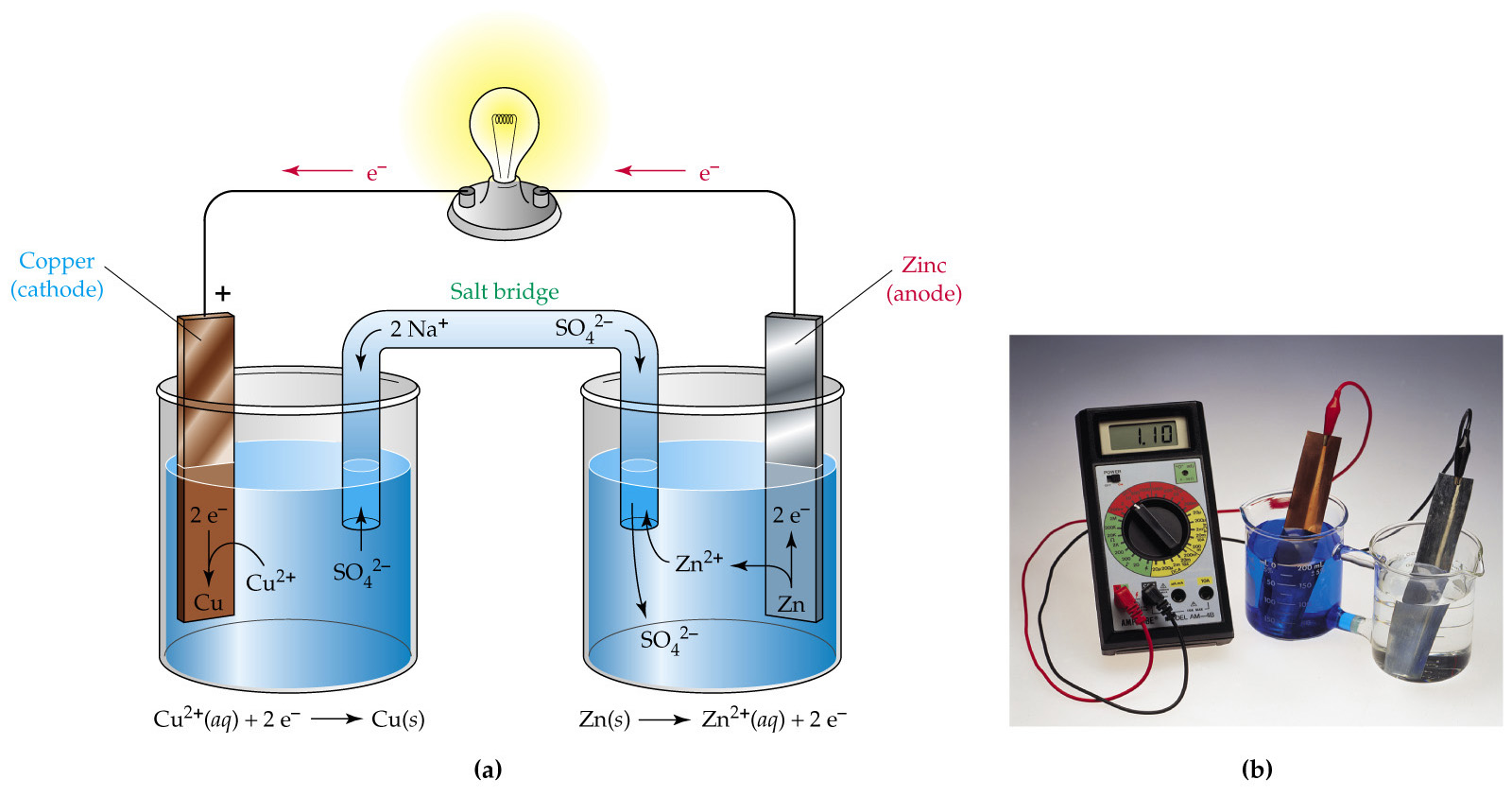
Polarographie
Définitions
La polarographie est une méthode électrochimique utilisant une électrode à goutte de mercure, qui permet d’analyser un très grand nombre d’espèces chimiques. Elle est essentiellement utilisée dans l’analyse des cations métalliques en solution aqueuse. Les limites de detection usuelles sont de l’ordre 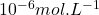 mais peuvent être portées jusqu’à
mais peuvent être portées jusqu’à 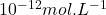 .
.
La polarographie, ainsi nommée par son inventeur nobélisé J. Heyrovsky, est à l’origine d’un grand nombre de techniques électroanalytiques dont la voltampérométrie. La polarographie est une méthode d’analyse qui consiste à étudier électrochimiquement des composés électroactifs en solution, au moyen d’une électrode indicatrice à goutte de mercure (goutte de mercure tombante ou goutte pendante, mais aussi film mince de mercure sur électrode solide). Elle se différencie de sa descendante, la voltampérométrie (utilisation des électrodes solides « platine, graphite, or… »), essentiellement par la nature de l’électrode puisque les méthodologies sont bien souvent identiques.
La polarographie regroupe un certain nombre de techniques analytiques couvrant un large domaine de concentrations allant de la polarographie classique, pour la moins sensible (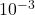 à
à 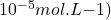 , aux méthodes de redissolution (adaptées aux traces et ultratraces) en passant par les méthodes impulsionnelles (
, aux méthodes de redissolution (adaptées aux traces et ultratraces) en passant par les méthodes impulsionnelles (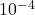 à
à 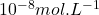 ). Les appareillages actuels permettent d’appliquer la plupart de ces techniques polarographiques. Elles sont applicables pour de nombreuses substances inorganiques, organiques, organométalliques ou biologiques dans un grand nombre de secteurs industriels et en recherche.
). Les appareillages actuels permettent d’appliquer la plupart de ces techniques polarographiques. Elles sont applicables pour de nombreuses substances inorganiques, organiques, organométalliques ou biologiques dans un grand nombre de secteurs industriels et en recherche.
La polarographie est une méthode indicatrice en régime stationnaire (sans agitation) utilisant
les courbes intensité-potentiel (courbes de polarisation) tracées en utilisant une électrode à gouttes de mercure. Le
transport en solution des espèces électroactives sur mercure est dû à la diffusion.
Dans les conditions de la polarographie :
- Les potentiels de demi-vague sont caractéristiques de la substance électoactive, d’où possibilité d’analyse qualitative;
- La hauteur des paliers est proportionnelle aux concentrations de ces substances, d’où possibilité d’analyse quantitative.
- Le domaine des potentiels accessibles sur mercure pour réaliser des réductions ou des oxydations électrochimiques s’étend, grosso modo,de +0,4 V à-2,0 V par rapport à l’électrode au calomel saturée (ECS)
- Le dispositif le plus utilisé est celui à 3 électrodes qui comprend une électrode de travail (la goutte de mercure), une électrode de référence et une électrode auxilliaire (platine)
Appareillage
La cellule de mesure en polarographie est classiquement constituée :
– d’un récipient qui contient la solution à analyser généralement en verre borosilicaté, d’un volume de 25 mL en général ;
– d’un support d’électrodes (couvercle) permettant l’étanchéité de l’ensemble et disposant d’un système d’arrivée de gaz pour assurer le dégazage de la solution et le maintien sous atmosphère inerte de la cellule ;
– d’un système d’agitation magnétique ou mécanique, afin d’homogénéiser la solution lors d’ajouts de réactifs ou de solutions étalons.
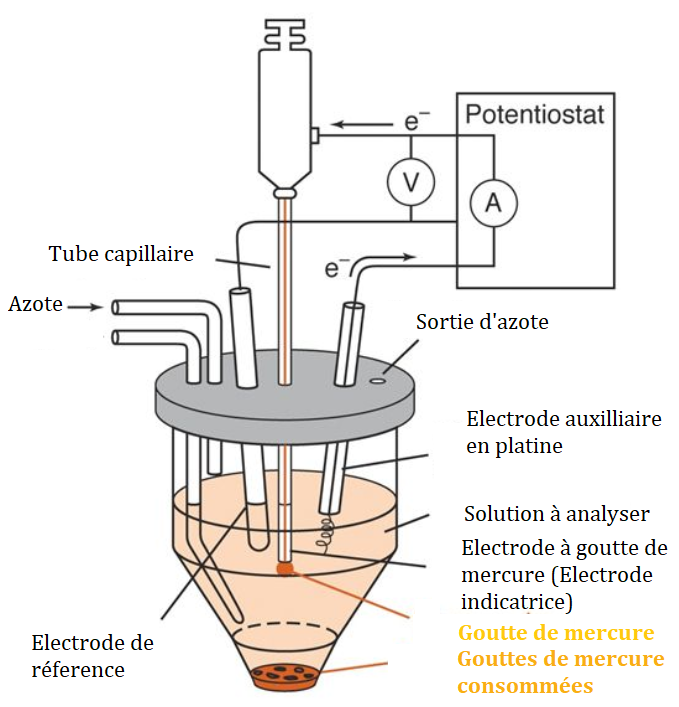
Les mesures polarographiques s’effectuent à partir d’un montage classique à trois électrodes.
- L’électrode de référence utilisée possède un potentiel constant et connu exactement. Même si l’électrode de référence au calomel saturée (ECS) est la plus utilisée, elle devrait, dans les années à venir, être supplantée par l’électrode de référence Ag/AgCI qui évite le recyclage d’une électrode en fin de vie contenant du mercure. Son potentiel sert de point de consigne au potentiostat.
- L’électrode indicatrice est constituée d’une électrode de mercure (DME, HMDE, TFME) dont le potentiel est imposé par le potentiostat par rapport à celui de l’électrode de référence.
- La contre-électrode est généralement constituée d’une tige de carbone vitreux ou d’un fil de platine de surface plus importante que l’électrode indicatrice de mercure (usuellement environ 50 fois plus grande).
Avantages de L’EGM
Les spécificités propres à l’électrode à goutte de mercure comparées aux électrodes conductrices solides viennent des propriétés particulières de ce métal qui, liquide à température ambiante, permet un renouvellement de la surface active de l’électrode, qui conduit aisément à la formation d’un certain nombre d’amalgames avec les métaux et qui permet des réductions à des potentiels très négatifs (réductions impossibles à réaliser sur électrode de platine ou de carbone vitreux). En oxydation, par contre, l’exploration des potentiels en polarographie est limitée par l’oxydation du mercure (+0,4 V), ce qui explique qu’une grande partie des applications a concerné des composés électroréductibles. Cette étendue des potentiels d’exploration est naturellement influencée par le pH, l’électrolyte support ou le solvant, ceux-ci pouvant limiter cette étendue.
Un autre avantage important du mercure réside dans le fait qu’à chaque goutte correspond une nouvelle électrode (Électrode renouvelable), identique à la précédente du point de vue géométrique, mais ne gardant pas mémoire du phénomène électrochimique ayant affecté les gouttes antécédentes, ce qui permet une bonne reproductibilité des analyses.
La faible surface de l’électrode entraine que l’intensité mesurée est très faible, de l’ordre de 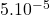 A. Il en résulte que les quantités de substances électrolysées sont infimes.
A. Il en résulte que les quantités de substances électrolysées sont infimes.
La surtension correspondant au dégagement d’hydrogène sur mercure est élevée ce gui signifie que l’apparition des vagues de réduction du proton et de l’eau se manifeste à des potentiels éloignés de leurs potentiels standard d’où un domaine large d’utilisation.
- Électrode renouvelable
- Bonne reproductibilité
- Domaine large (Hg
 surtension avec l’hydrogène)
surtension avec l’hydrogène) - Avec Hg mur reculé (analyse de plusieurs espèces)
- Zone de concentration:
- Classique :
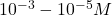
- Impulsionnelle :
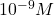
- Classique :
Méthode de spéciation des formes organométalliques.
Les solutés à étudier parviennent à la surface de l’électrode par diffusion pure. Le transport par migration est exclu par addition systématique d’un électrolyte support (électrolyte indifférent) non électroactif et au moins 100 fois plus concentré que l’espèce à analyser.
L’inconvénient majeur que présente l’électrode à gouttes de mercure se situe au niveau des oxydations. Le domaine des oxydations possibles est peu étendu.
Au-dessus de +0,4 V, le mercure s’oxyde en 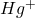 en donnant une vague anodique. L’électrode se dissout. C’est la raison pour laquelle la polarographie est peu utilisée en oxydation. Si, toutefois, on veut réaliser des déterminations par oxydation, d’autres microélectrodes polarisables telles qu’un fil de platine tournant ou des électrodes stationnaires solides (platine, or, graphite…) sont utilisables (voltamétrie).
en donnant une vague anodique. L’électrode se dissout. C’est la raison pour laquelle la polarographie est peu utilisée en oxydation. Si, toutefois, on veut réaliser des déterminations par oxydation, d’autres microélectrodes polarisables telles qu’un fil de platine tournant ou des électrodes stationnaires solides (platine, or, graphite…) sont utilisables (voltamétrie).
Téchniques Polarographiques
Les techniques polarographiques couvrent un large domaine d’applications en analyse et peuvent être utilisées en particulier dans les domaines de l’environnement, de l’analyse de l’eau et des eaux résiduaires, de l’industrie pharmaceutique, alimentaire, cosmétique, pétrolière et nucléaire, de la galvanoplastie, de l’analyse des fluides biologiques et de l’industrie des plastiques et des polymères. Elles sont appliquées à l’analyse de
constituants principaux et à l’analyse de traces et d’ultratraces. Elles concernent l’analyse en solution de métaux, de molécules inorganiques, organométalliques ou organiques ou des molécules d’intérêt biologique. Elles permettent des analyses multiélémentaires et sont non destructives des solutions analysées puisque les quantités de solutés impliqués dans les mesures polarographiques sont négligeables par rapport aux quantités en solution, permettant ainsi des mesures répétitives sur une même solution. Elles nécessitent quelques millilitres à une cinquantaine de millilitres de solution selon la taille de la cellule électrochimique utilisée et peuvent être appliquées, soit directement sur l’échantillon lorsque c’est une solution, avec éventuellement une adaptation de ses conditions chimiques (ajout d’un électrolyte support, modification du pH, ajout d’un complexant) ou un ajustement des concentrations avec une étape de dilution, soit après une étape de prétraitement de l’échantillon (filtration, extraction, digestion, oxydation, solubilisation, dissolution, purification) pour des matrices complexes ou solides comme c’est le cas pour de nombreuses autres méthodes d’analyse.
En pratique, il résulte de l’application des méthodes d’analyse polarographiques la mesure d’un courant ou parfois d’une quantité de charges, à un potentiel caractéristique de l’analyte, proportionnel à sa concentration en solution. Pour les analyses quantitatives, on procède, soit à l’établissement d’une droite d’étalonnage à partir de concentrations connues du soluté à laquelle on se reporte pour la détermination d’une concentration inconnue, soit pour éviter des erreurs de mesures dues aux effets de matrices, à la méthode des ajouts dosés, en effectuant des ajouts de concentrations connues dans la solution contenant le ou les solutés à analyser.
Les différentes techniques polarographiques couramment utilisées seront décrites ci-après présentant le principe de leurs mises en œuvre, leurs performances et les applications qui les concernent illustrées de quelques exemples pratiques. Ainsi seront abordées la polarographie classique et les techniques de polarographie impulsionnelle qui utilisent une électrode à goutte de mercure tombante (DME ou SMDE) et qui sont adaptées au domaine de concentration allant des centaines de mg · L–1 à la centaine de µg · L–1. Pour les analyses de traces et d’ultratraces, les méthodes de redissolution anodique et cathodique utilisant une électrode de mercure statique (HMDE, TFME) seront également présentées.
Polarographie classique
Polarographie classique regroupe la polarographie à courant direct (DCP), introduite par Heyrovsky en 1923, et la polarographie à courant direct échantillonné (appelée aussi Tast Polarography, DCTP). À partir d’un montage à trois électrodes, elle est caractérisée par l’enregistrement de courbes intensité-potentiel dans des solutions non agitées avec une électrode à goutte de mercure tombante (DME) comme électrode de travail.
Polarographie à courant direct (DCP)
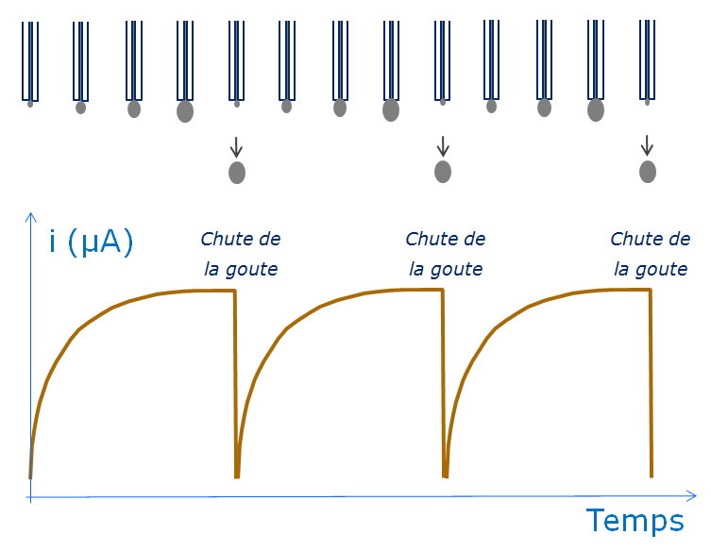
En faisant varier linéairement le potentiel à l’EGM à faible vitesse de balayage des potentiels 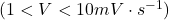 , le polarogramme enregistré en DCP présente des oscillations causées par la croissance et la chute de la goutte à intervalle de temps régulier (
, le polarogramme enregistré en DCP présente des oscillations causées par la croissance et la chute de la goutte à intervalle de temps régulier (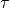 ). Le signal obtenu pour une espèce qui se réduit à l’EGM (comme c’est souvent le cas en polarographie) se présente sous la forme d’une vague sigmoïde.
). Le signal obtenu pour une espèce qui se réduit à l’EGM (comme c’est souvent le cas en polarographie) se présente sous la forme d’une vague sigmoïde.
La figure suivante représente un exemple de polarogramme obtenu en DCP correspondant à la réduction de l’acide pyruvique. La hauteur de cette vague correspond au courant limite de diffusion (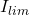 ) ( voir l’équation d’Ilkovic) :
) ( voir l’équation d’Ilkovic) :
(1) 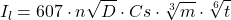
Dans cette équation, le courant est en ampères si l’on exprime le débit de mercure, 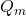 , en grammes par seconde, le temps de goutte en secondes, le coefficient de diffusion, D, en centimètres carrés par seconde et la concentration de l’espèce en solution qui se réduit,
, en grammes par seconde, le temps de goutte en secondes, le coefficient de diffusion, D, en centimètres carrés par seconde et la concentration de l’espèce en solution qui se réduit, 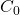 , en moles par centimètre cube.
, en moles par centimètre cube.
Comme l’indique l’équation d’Ilkovic, à débit de mercure constant, le courant de diffusion est directement proportionnel à la concentration molaire de l’espèce électroactive en solution, permettant ainsi d’utiliser la polarographie en analyse quantitative.

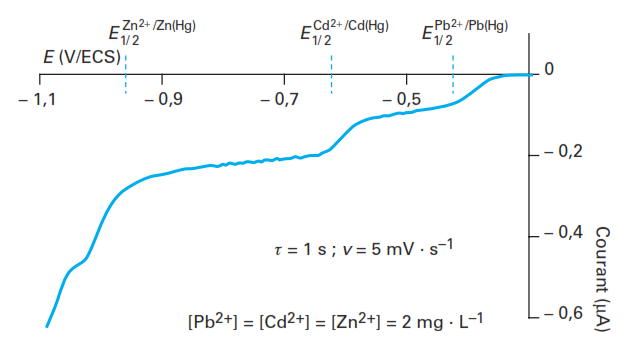
Les courants de diffusion provenant de plusieurs espèces électroactives sont additionnels comme le montre la figure suivante dans le cas de l’analyse d’un mélange plomb-cadmium-zinc avec trois vagues de réduction successives.
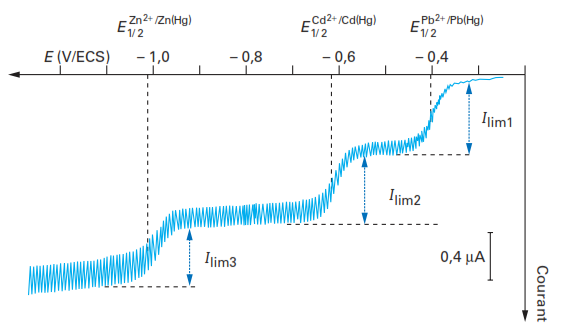
Remarques: le potentiel de demi-vague d’une espèce électroactive peut être soumis à variations selon les systèmes électrochimiques mis en jeu et les conditions chimiques correspondant à l’électrolyte support.
Polarographie à courant direct échantillonné (Tast polarography DCTP)
Afin d’éliminer les oscillations de courant dues à la croissance de la goutte de mercure et sa chute, et d’avoir un rapport courant faradique/courant capacitif le plus important possible, le courant peut être enregistré uniquement à la fin de la vie de la goutte pendant un temps faible (tm) de 1 à 100 ms (figure a ci-dessous ).

Dans ce cas, il est nécessaire d’avoir une parfaite coïncidence entre l’échantillonnage du courant et la chute de la goutte qui est provoquée artificiellement avec un système de frappe du capillaire. C’est ce qui a donné lieu à la technique de polarographie à courant direct échantillonnée (Tast Polarography, DCTP). Entre les échantillonnages, le courant est maintenu à la dernière valeur mesurée.
Le polarogramme obtenu se présente sous la forme de courbes sigmoïdes qui ne présentent plus d’oscillations (figure ci-dessous) et dont les caractéristiques sont les mêmes que celles décrites précédemment en DCP (pour Ilim et E1/2).
Le contrôle numérique des appareillages de polarographie a conduit à remplacer la variation linéaire de potentiel par une rampe incrémentée de potentiel. Les incréments de potentiel (∆Es) sont imposés tous les temps ts qui coïncident avec le temps de goutte (figure ). Le courant est alors échantillonné à la fin de la
vie de la goutte sur une période très courte ™ pour considérer la surface de l’EGM pratiquement constante et réduire ainsi la composante capacitive du courant total mesuré.
Une autre amélioration de la technique de polarographie classique permettant d’améliorer le rapport if /ic est l’utilisation de l’électrode à goutte tombante de mercure en mode statique (SMDE) (figure ) grâce à l’utilisation d’électrodes à gouttes de mercure multimodes . Dans ce cas, au bout d’un certain temps par rapport au début de la croissance de la goutte de mercure, le débit de mercure est interrompu et la surface de la goutte devient alors constante permettant ainsi l’échantillonnage du courant en fin de vie de la goutte après une décroissance notable du courant capacitif (la surface de la goutte ne variant plus).
Polarographie classique est généralement appliquée pour l’analyse de solutés se situant dans le domaine de concentration compris entre 5 × 10–5 et 5 × 10–3 M. C’est l’existence d’un courant capacitif (ic) dû à la charge et la décharge de la double couche électrique à l’interface électrode-solution qui impose la limite de détection de la polarographie classique. En effet, le courant enregistré tout au long de la vie de la goutte de mercure est la somme de deux composantes : le courant capacitif (ic) et le courant faradique (if) correspondant au signal analytique. Ces deux courants dépendent du temps et varient en sens opposés au cours de la variation de la surface de l’EGM :
- le courant capacitif varie en fonction de t–1/3 dans le cas où le potentiel de l’électrode pendant la durée de vie de la goutte a une variation négligeable [faibles vitesses de balayage des potentiels (< 10 mV · s–1)] ;
- le courant faradique varie, lui, en fonction de t1/6.
À la fin de la vie de la goutte, le rapport if /ic est alors maximal. Pour des concentrations en analyte dans l’intervalle 5 × 10–5– 10–2M, le courant enregistré est essentiellement faradique (ic est négligeable) et une vague bien définie est obtenue sur le polarogramme. Pour des niveaux de concentration plus faibles, le courant capacitif devient comparable au courant faradique et la mesure du courant limite de la vague devient difficile. C’est donc le courant capacitif qui limite en DCP la détection d’analytes à des concentrations comprises entre 5 × 10–6 et 10–5M.
La méthode polarographique à courant direct échantillonné permet d’améliorer légèrement le rapport if /ic et donc les limites de détection de la polarographie classique, généralement d’un facteur de 2 à 5. Ainsi par exemple, Bond et Canterford ont montré que la limite de détection du cuivre passe de 3 × 10–6M en DCP à 1 × 10–6 M en DCTP .
Polarographie impulsionnelle
Les méthodes impulsionnelles ont été développées dans les années 1960 [59] [60] afin de pallier le problème de sensibilité de la polarographie classique dû à la composante capacitive du courant mesuré qui ne permet pas de l’utiliser pour des concentrations nettement inférieures à 10–5M. Plusieurs méthodes ont vu le jour et se différencient par le mode d’application du potentiel à l’électrode de mercure. Les techniques présentées ici sont celles qui sont couramment employées, c’est-à-dire :
- la polarographie à impulsions normales (NPP) ;
- la polarographie à impulsions constantes ou polarographie impulsionnelle différentielle (DPP) ;
- la polarographie impulsionnelle à tension carrée (SWP).
En polarographie impulsionnelle, l’application du potentiel pour ces techniques s’effectue avec une électrode à goutte de mercure tombante (DME ou SMDE) dont la chute est provoquée mécaniquement à un temps prédéfini ou à une électrode de mercure stationnaire (HDME, TFME). Lorsque le mode impulsionnel s’applique à d’autres électrodes que le mercure (par exemple électrodes à disques de graphite, de platine…) le terme voltampérométrie impulsionnelle doit être employé.
Ainsi, avec l’électrode à goutte de mercure tombante, l’impulsion de potentiel s’effectue sur un temps court devant le temps de vie de la goutte, là où sa surface est maximale (fin de la vie de la goutte).
Polarographie impulsionnelle normale ou polarographie à impulsions croissantes (NPP)
Dans ce cas, le potentiel de l’électrode de travail ne varie pas linéairement avec le temps comme en polarographie classique mais est maintenu à un potentiel initial E constant choisi généralement de telle façon qu’aucune réaction électrochimique ne se produise.
Ainsi, lors d’une réduction, le potentiel Ei sera beaucoup moins négatif que le E1/2 du système redox étudié. Comme le montre la figure 8, des impulsions croissantes sont surimposées à E.

Elles sont synchronisées avec le temps de goutte et appliquées juste avant la fin de vie de la goutte. À la fin de chaque impulsion, le potentiel revient au potentiel initial Ei. Une seule impulsion de potentiel est appliquée à chaque goutte pendant une durée ti de 30 à 100 ms et son amplitude  augmente d’une impulsion à l’autre avec un incrément en général faible de 1 à 6 mV pour atteindre un maximum de 1000 mV. En effet, pour avoir une bonne définition des vagues, il faut un nombre de points suffisant, donc un assez grand nombre d’impulsions sur l’intervalle de potentiel balayé. De plus, la durée d’analyse étant limitée, il est usuel d’employer des vitesses apparentes de balayage de 1 à 5 mV.s-1. Les vitesses supérieures sont proscrites.
augmente d’une impulsion à l’autre avec un incrément en général faible de 1 à 6 mV pour atteindre un maximum de 1000 mV. En effet, pour avoir une bonne définition des vagues, il faut un nombre de points suffisant, donc un assez grand nombre d’impulsions sur l’intervalle de potentiel balayé. De plus, la durée d’analyse étant limitée, il est usuel d’employer des vitesses apparentes de balayage de 1 à 5 mV.s-1. Les vitesses supérieures sont proscrites.
Polarographie impulsionnelle différentielle (DPP)
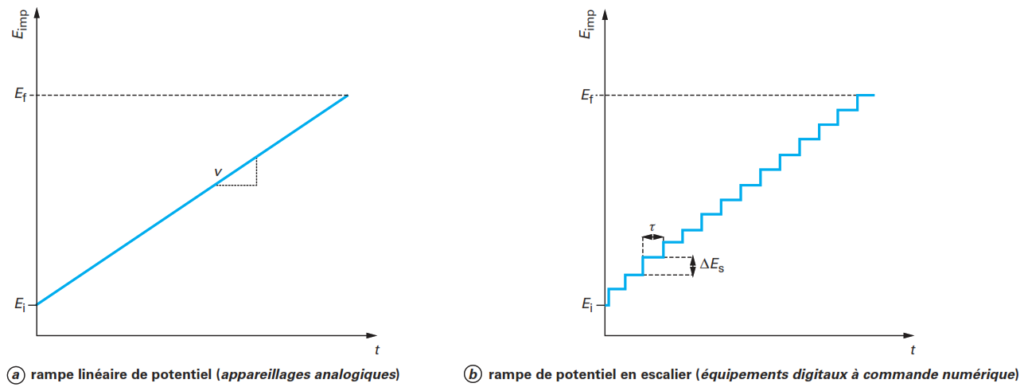
Dans le cas de la polarographie impulsionnelle différentielle, appelée encore polarographie à impulsions constantes, les appareillages commerciaux délivrent les deux types de programmations schématisées sur la figure 10 où une impulsion de potentiel DE de faible amplitude (10 à 100 mV) est surimposée à une rampe linéaire de potentiel variant lentement à la vitesse de balayage v (cas des appareillages analogiques) ou bien à rampe de potentiel en escalier assurant la vitesse de balayage v avec un potentiel incrémenté d’un échelon de potentiel (DEs) tous les temps ts égaux au temps de goutte t (cas des appareillages numériques). Dans les deux cas, cette impulsion DE est appliquée pendant un temps court tp de 10 à 100 ms à la fin de la vie de la goutte juste avant sa chute provoquée par un dispositif mécanique asservi de frappe du capillaire.
Les vitesses de balayage du potentiel utilisées sont classiquement comprises entre 1 et 10 mV · s–1 et les temps de goutte de 0,5 à 3 s.
En DPP, le courant est échantillonné deux fois pendant le temps de vie de la goutte, comme indiqué sur la figure 11e, juste avant l’application de l’impulsion de potentiel (i1) et à la fin de l’impulsion (i2) sur de courtes périodes (tm1 et tm2 compris entre 5 et 20 ms). Le signal enregistré est la différence ∆i entre ces deux courants (figure 11f ). Cette valeur est conservée jusqu’à la mesure suivante (nouvelle goutte) et le polarogramme est tracé en portant ∆i en fonction du potentiel de base appliqué (rampe de variation linéaire du potentiel) et se présente sous forme de petites « marches d’escalier » successives dont la longueur du palier correspond à τ.
En présence d’une substance électroactive et lorsque le potentiel imposé est suffisamment négatif pour qu’elle soit réduite à l’électrode à goutte de mercure, le courant produit pendant le temps de vie de la goutte peut être décomposé en trois composantes principales:
- une composante de courant faradique continu (figure11b) dû à la variation du potentiel et à la croissance de la surface de la goutte comme décrit précédemment en polarographie classique et qui croît en fonction de t1/6 ;
- une composante capacitive créée par l’application de l’impulsion de potentiel ∆E correspondant au courant capacitif transitoire trans ic qui décroît exponentiellement en fonction du temps (figure 11c ) ;
- une composante faradique due à l’impulsion de potentiel correspondant au courant faradique transitoire iftrans qui décroît en fonction de t–1/2 (figure 11d ).
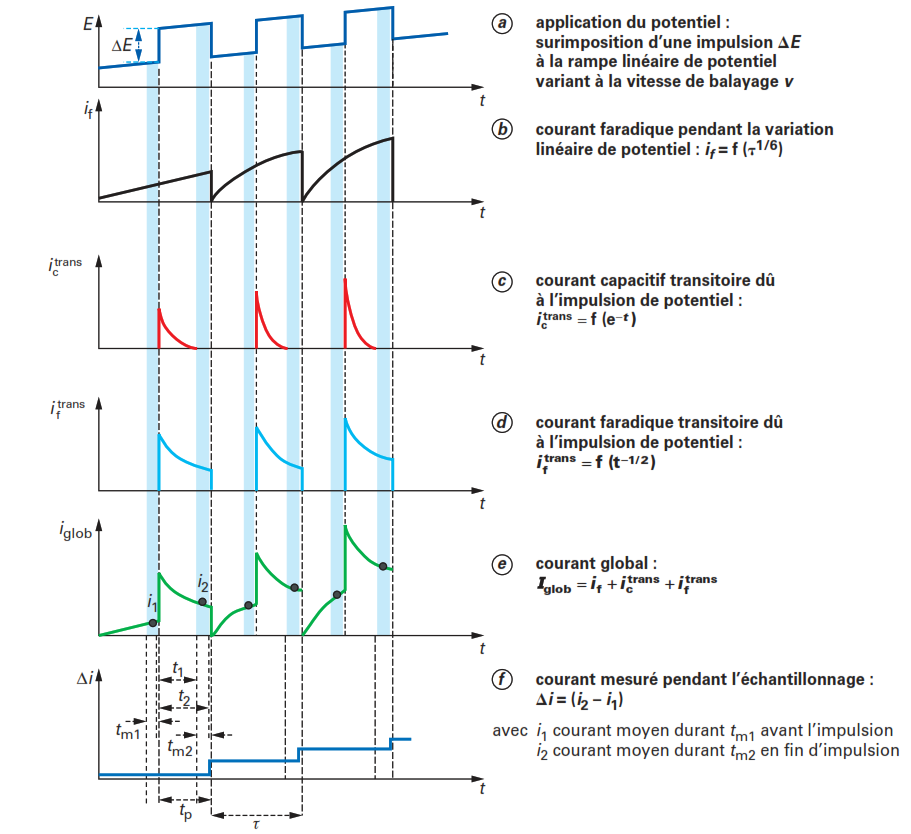
Le courant global, iglob , correspond à la somme de ces trois composantes (figure 11e ). La mesure du courant à la fin de l’impulsion (entre t1 et t2) permet ainsi de rendre négligeable la composante ictrans dans iglob et la mesure de ∆i conduit à ne mesurer pratiquement que la variation de courant faradique due à l’impulsion. Ainsi, la sensibilité en DPP se trouve fortement améliorée par rapport à la polarographie classique.
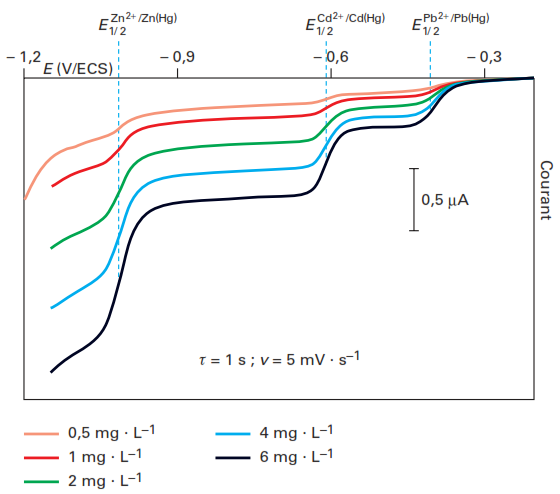
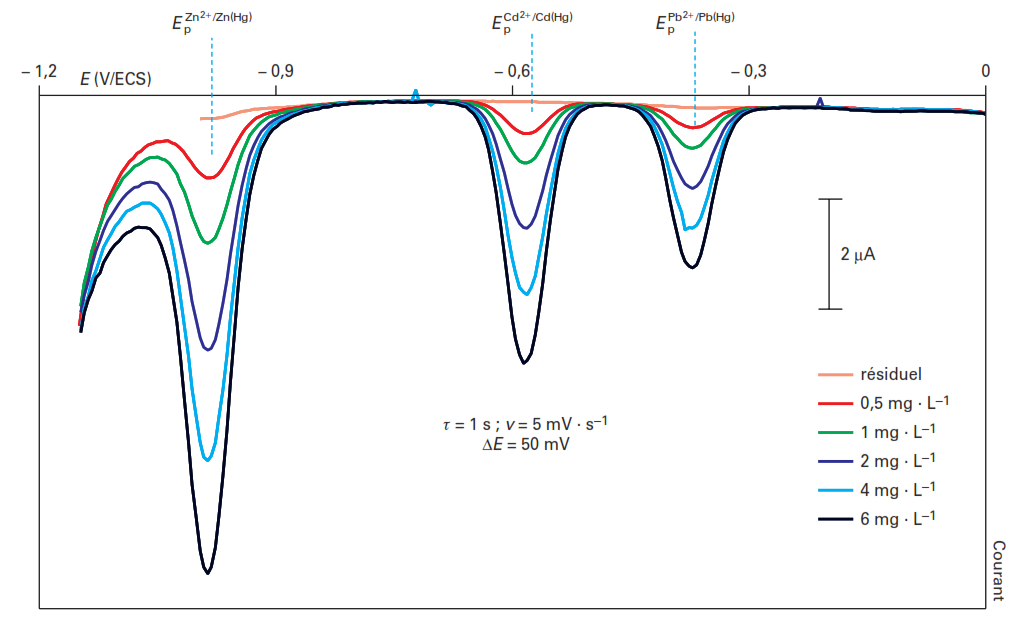
Le polarogramme obtenu pour une substance électroactive présente, compte tenu de la mesure par échantillonnage et différentiation, est une courbe sous forme d’un pic symétrique. À titre d’exemple, la figure ci-dessous présente les polarogrammes en DPP pour un mélange plomb-cadmium-zinc en fonction de la concentration
des cations en solution faisant apparaître trois pics de réduction successifs qui correspondent respectivement à la réduction de Pb2+ , Cd2+ et Zn2+.
HCl 0,1 M
Voltamétrie à redissolution
Les techniques d’analyse polarographiques par redissolution (stripping polarography) sont des techniques extrêmement sensibles pour l’analyse de traces et d’ultratraces .
Cette sensibilité remarquable est attribuée à la combinaison d’une étape de préconcentration au niveau d’une électrode de mercure associée à une étape de mesure qui consiste en une étape de redissolution. Il est ainsi possible d’effectuer des analyses mono et multi-élémentaires (pouvant atteindre simultanément 5 à 6 analytes) dans différentes matrices aqueuses. Ce sont des techniques qui s’appliquent sans étape de concentration chimique en amont de l’étape de préconcentration électrochimique et qui présentent des limites de détection de 10-10 à 10-11mol.L-1 pouvant atteindre 10-12mol.L-1 pour certains métaux, ce qui rend ces méthodes parmi les plus sensibles des méthodes d’analyse existantes.
L’avantage des méthodes polarographiques par redissolution vient du fait que les deux étapes préconcentration-mesure s’effectuent dans la même solution analysée et qu’il est possible de répéter les mesures sans altération de celle-ci.
Selon le processus d’accumulation à l’électrode de mercure (HMDE ou TFME) au cours de la première étape (dépôt de l’analyte électrochimiquement sous forme métallique ou de sel insoluble ou sous forme adsorbée) et selon la méthode de redissolution utilisée, plusieurs techniques électrochimiques par redissolution ont été développées et les principales d’entre elles seront décrites dans les paragraphes suivants.
Les techniques d’analyse polarographiques par redissolution sont des techniques d’analyse qui n’affectent pas la composition des solutions analysées et qui s’effectuent essentiellement en deux étapes principales :
- une première étape de dépôt à l’électrode de mercure stationnaire (HMDE ou TFME)sous agitation, allant de trente secondes à quelques minutes, au cours de laquelle une très faible proportion de l’analyte en solution est déposée et préconcentrée à l’électrode d’un facteur 100 à 1 000 permettant ainsi des limites de détection très faibles ;
- une seconde étape de redissolution (stripping ) par oxydation ou réduction du ou des solutés déposés préalablement, celle-ci étant une étape de mesure proprement dite et utilisant généralement une des méthodes polarographiques décrites dans les paragraphes précédents (variation du potentiel en fonction du temps en mode linéaire ou impulsionnel et mesure du courant) ou une méthode chronopotentiométrique (mesure du potentiel en fonction du temps ).
Entre ces deux étapes principales vient dans la plupart des cas s’intercaler une étape dite de repos où la solution n’est plus agitée pendant 20 à 30 s. Cette étape, lorsqu’il y a formation d’un amalgame entre le métal déposé et le mercure permet d’obtenir une homogénéité de l’amalgame dans la goutte ou le film de mercure.
Elle permet également de s’assurer que la convection de la solution est limitée à la convection naturelle puisque la solution est agitée au cours de la première étape.
Selon le processus de dépôt à l’électrode de mercure au cours de l’étape 1 et de la technique polarographique employée pour l’étape 2 de redissolution, différentes techniques particulières de polarographie par redissolution peuvent être mises en œuvre. Elles sont résumées dans le tableau 4 qui présente pour chacune d’elles les processus de dépôt et les techniques de mesure employées dans l’étape de redissolution. Ces différentes techniques et leurs domaines d’application sont présentés plus en détail dans les paragraphes suivants.
Ainsi, lorsqu’une technique polarographique à variation de potentiel est utilisée pour l’étape de redissolution, la technique est appelée, pour une variation de potentiel vers les potentiels anodiques, polarographie par redissolution anodique (ASP pour Anodic Stripping Polarography ) et, pour un balayage s’effectuant dans le sens cathodique, polarographie par redissolution cathodique (CSP pour Cathodic Stripping Polarography). La méthode de polarographie par redissolution anodique est utilisée principalement pour l’analyse de métaux formant un amalgame avec le mercure alors que la méthode de redissolution cathodique est utilisée pour déterminer des solutés qui forment un complexe insoluble à la surface de l’électrode avec le mercure oxydé.
Polarographie par redissolution anodique (Anodic Stripping Polarography )
Les techniques de polarographie par redissolution anodique combinent une étape de préconcentration d’un ou plusieurs ions métalliques à analyser par électrodépôt à potentiel cathodique contrôlé au niveau de l’électrode indicatrice de mercure (HMDE ou TFME) et une étape suivante où le potentiel varie et qui correspond à un processus de dissolution anodique où le métal préconcentré est oxydé.

Au préalable réduire la totalité de l’élément présent sur la goutte de Hg
Réduction (Hg = cathode)
- E cte < E1/2 = préélectrolyse.
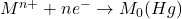
- Redissoudre l’amalgame en faisant défiler les E vers les valeurs (+)
Oxydation (Hg devient anode)
La redissolution commence lorsqu’on s’approche de E1/2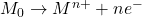
- En goutte de mercure pendante :Dosage Zn, Cd, Cu, Pb
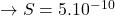 M
M - En électrode solide (Au, Pt, Ag) : Dosage As
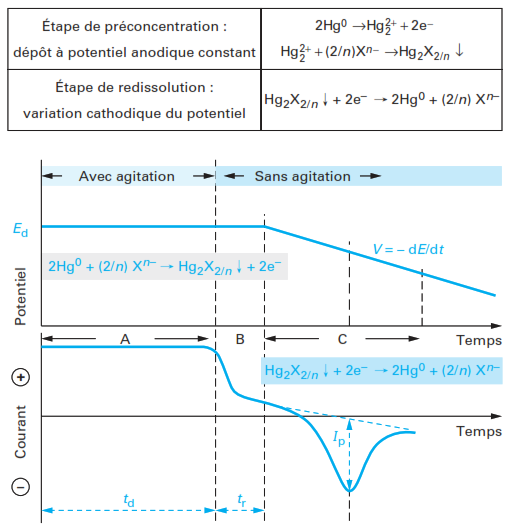
Polarographie par redissolution cathodique (Cathodic Stripping Polarography )
À l’origine, la polarographie par redissolution cathodique a été utilisée pour l’analyse d’anions inorganiques ou organiques et diffère de la technique de polarographie par redissolution anodique, par la méthode de redissolution, mais surtout par le processus d’accumulation associé. Les réactions électrochimiques et chimiques impliquées dans ces deux étapes peuvent être résumées sur le schéma suivant.
Difficultés particulières de la méthode
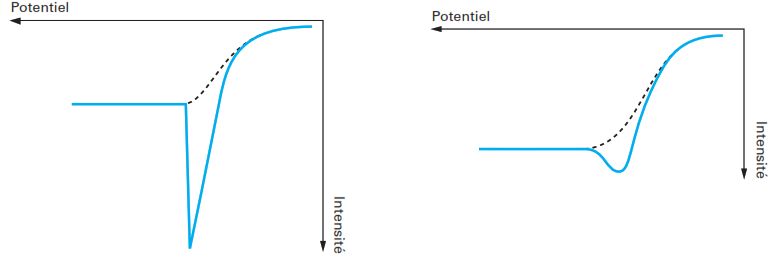
Effets de maxima polarographiques
Parmi les phénomènes qui peuvent modifier les vagues polarographiques, il existe certaines déformations très particulières qui ont reçu le nom de maximums car elles conduisent à des courants plus élevés que les courants attendus en certains points du polarogramme (figure suivante).
Ce phénomène est en général du à des mouvements anormaux de la solution au voisinage immédiat de l’électrode, entraînant des variations brutales du flux de l’espèce électroactive, donc du courant.
La présence d’espèces tensioactives permet d’atténuer ou de supprimer ce phénomène.
Le facteur qui semble être aussi important est le débit du mercure dans les solutions concentrées:
le maximum n’apparaît qu’aux débits élevés (supérieurs à 2.10-3g.s-1) lorsque la durée de vie de goutte est à d’environ une seconde. Le courant n’est plus contrôlé par la diffusion, mais aussi par la convection.
En analyse polarographique, l’apparition d’un maximum est en général un inconvénient auquel il faut remédier. L’addition de gélatine a été d’abord proposée, mais la stabilité dans le temps du Triton X 100 le fait souvent préférer.
L’addition des suppresseurs doit se faire en très faibles proportions, car si le maximum est supprimé, on constate que le courant limite subit aussi une diminution pouvant atteindre 20 %. En général, une valeur de 10–3 % en agent suppresseur (gélatine, Triton X 100, empois d’amidon, rouge de méthyle …) peut suffire.
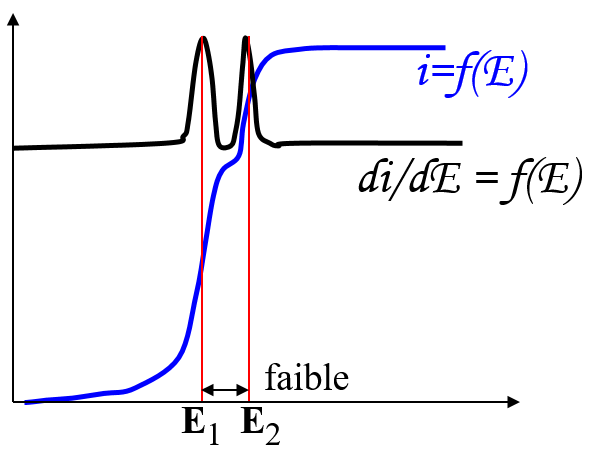
Problème des espèces successives
Différence de concentration énorme
→ les hauteurs très différentes
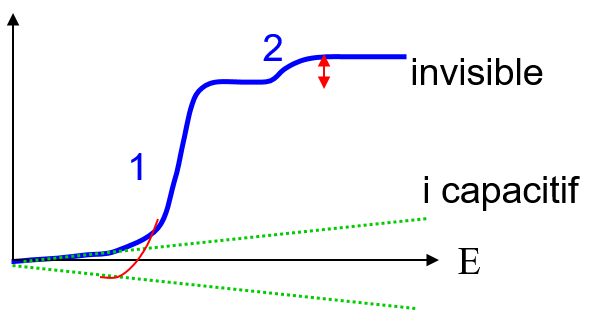
les E1/2 proches E1 ≈ E2
Réduction de O2
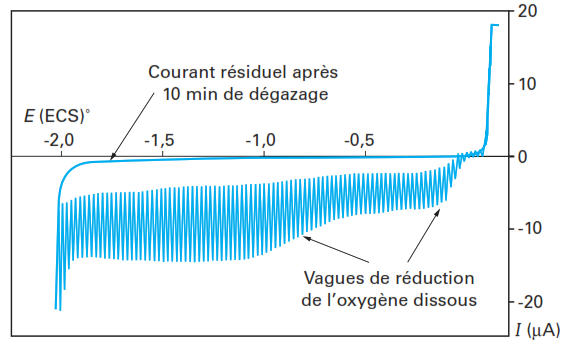
Le courant résiduel, qui correspond au courant obtenu au cours du tracé d’un polarogramme d’une solution électrolytique ne contenant pas d’espèces électroactives intentionnellement ajoutées:
- est liée à la réduction ou à l’oxydation de traces d’impuretés qui sont presque inévitables et qui proviennent généralement d’ions de métaux lourds et de diverses impuretés présentes dans les sels utilisés comme électrolyte support et dans le solvant (utiliser des produits de grande pureté)
- essentiellement à l’oxygène présent dans la solution à analyser. Pour chasser l’oxygène dissous de la solution un dégazage suffisants d’azote (10 min environ) rend presque négligeable cette composante du courant résiduel.
En milieu acide :
(2) 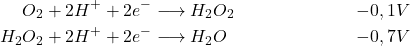
En milieu basique :
(3) 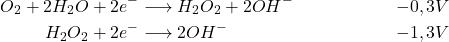
Voltamétrie cyclique (A.S.V)
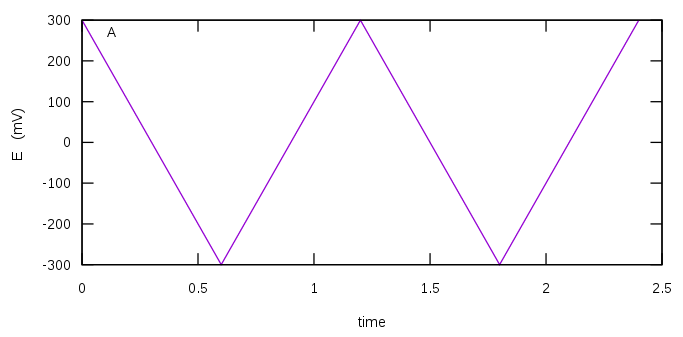
Le principe de la Voltamétrie cyclique (ASV) consiste à appliquer à une petite électrode de travail stationnaire (platine « voltamétrie », mercure « polarographie ») une tension variant linéairement avec le temps.Le balayage de tension s’effectue à partir d’une tension dite de base E_b}, où aucun transfert de charge ne s’effectue,jusqu’à une tension finale E_f}, ou une oxydation ou une réduction s’effectue ou s’est déjà produite. Après la traversée de la région de potentiel où l’espèce étudiée est électroactive, le sens du balayage est inversé. On effectue alors, à la même vitesse, le balayage de E_f} à E_b}, c’est le balayage retour. Plusieurs cycles successifs peuvent être réalisés. Le profil du potentiel imposé à l’électrode en fonction du temps est donné par la figure ci-dessous.
La figure suivante montre la morphologie du voltampérogramme obtenu avec un composé ayant le comportement d’un système rapide (réversible).
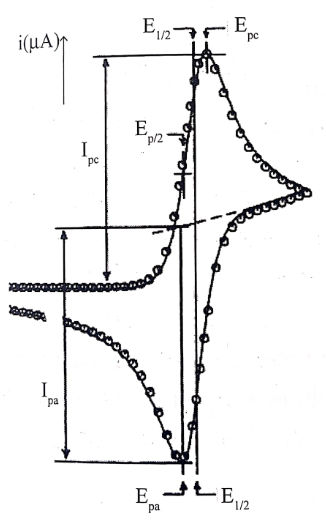
La figure suivante montre le cas d’un système lent (irréversible), ou les potentiels de pics de réduction (E_{pc}}) et d’oxydation (E_{pa}}) s’écartent beaucoup l’un de l’autre que dans le cas des systèmes rapide .

Possibilités analytiques
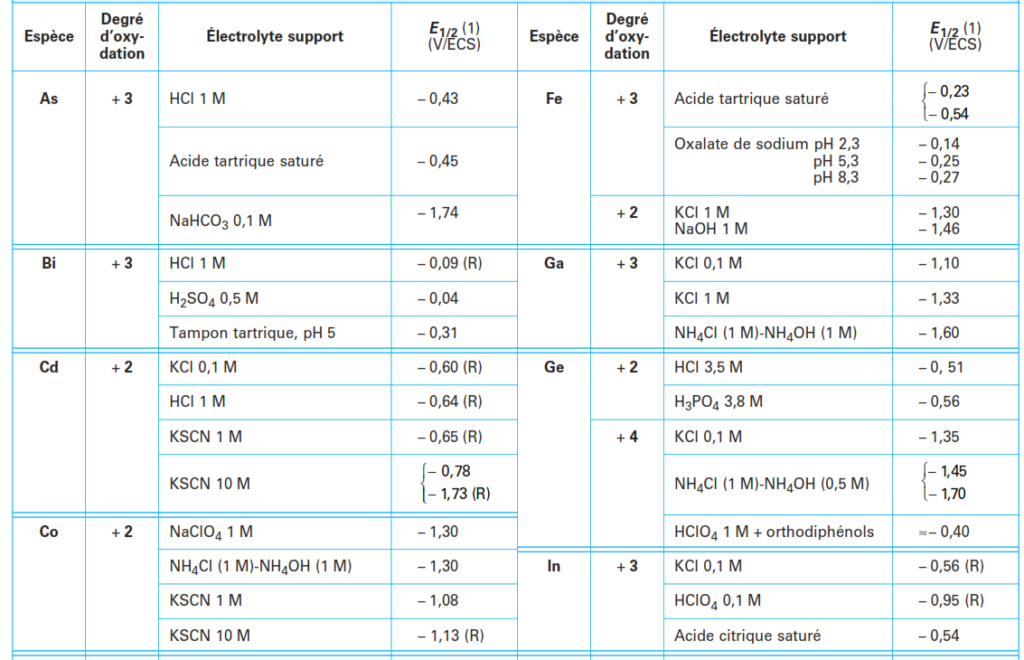
- Qualitative: la polarogaphie permet d’identifier les substances presentes dans la solution en comparant leurs potentiels de demi-vague avec ceux mentionnées dans des tables de potentiel de demi-vague en fonction de l’électrolyte utilisé.
- Quantitative : Absolue par l’équation d’ILKOVIC, mais en pratique on utilise les méthodes d’étalonnage:
Comparaison directe :
- Étalon:
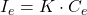
- Échantillon:
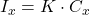
Droite d’étalonnage :
On prépare une série de solutions à différentes concentrations de l’analyte, l’électrolyte support et un suppresseur de maxima étant ajoutés dans les mêmes proportions pour les étalons que pour la solution à analyser (échantillon Cx).
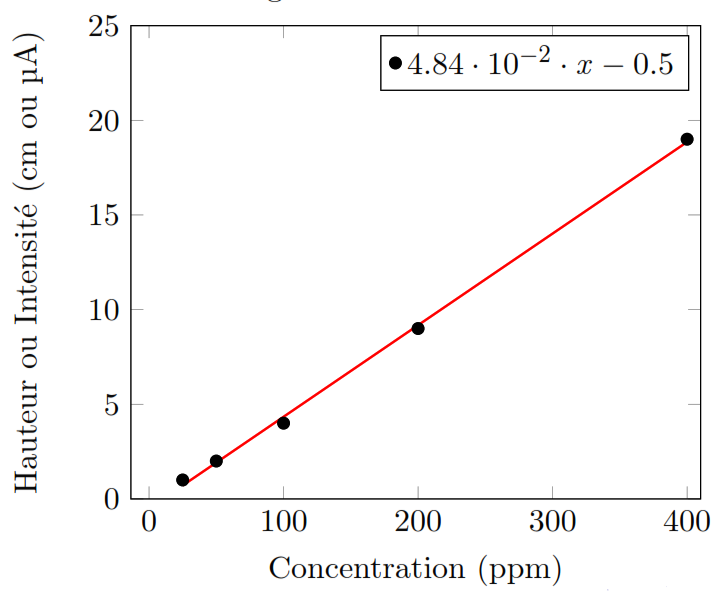
Méthode des ajouts dosées
Un seul ajout
Ainsi, après avoir appliqué la méthode à un volume connu exactement de l’échantillon analysé conduisant à l’enregistrement d’un premier polarogramme, un micro-ajout d’une solution standard du métal à doser est introduit dans la solution échantillon. Après dégazage et homogénéisation, la méthode est de nouveau appliquée pour donner un nouvel enregistrement.
Il est nécessaire que la quantité de standard ajouté soit suffisante pour que le courant de pic augmente de façon significative (la valeur du courant ait au moins doublé).
1
\begin{align}i_x&= K \times C_x \\ i_x &= \text{intensité de l’échantillon}\\C_x &= \text{la concentration inconnue de l’échantillon}\end{align}
2
\begin{align} i_e &= K \times \dfrac{(V_x \times C_x) + (V_e \times C_e) }{V_x + V_e} \\V_e&= \text{le volume de l’étalon ajouté} \\V_x&= \text{le volume de l’échantillon initial}\\ C_e&= \text{la concentration connue de l’étalon ajouté} \\i_e&= \text{intensité de la solution après premier ajout} \end{align}
\begin{align*} K&= i_e \times \dfrac{(V_x + V_e)}{(V_x \times C_x) + (V_e \times C_e) } \\ \text{Puisque}: \qquad C_x &= \dfrac{i_x}{K}\\ C_x &=i_x \times \dfrac{\left[(V_x \times C_x) + (V_e \times C_e) \right]}{i_e\times (V_x + V_e)}\end{align*}
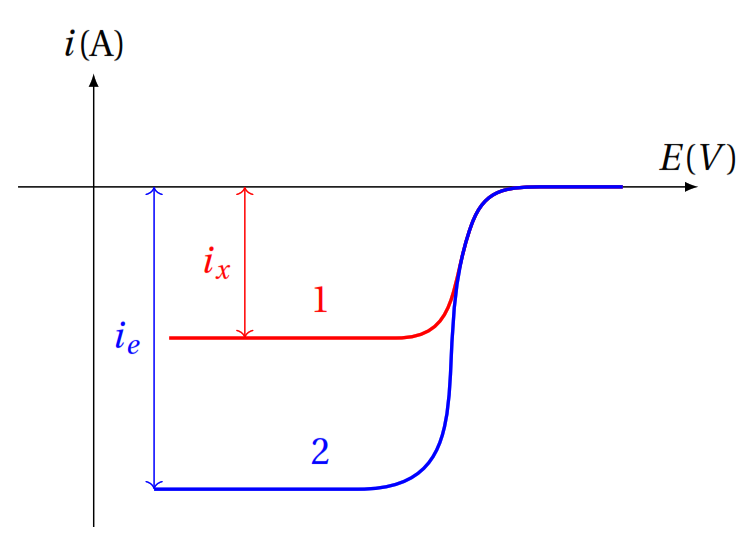
Ajouts multiples
Pour augmenter la précision de cette méthode; plusieurs ajouts successifs (idéalement au moins 3) doivent être effectués avant d’exploiter la courbe, l’équation de la droite obtenue (figure ??) permet de déterminer la concentration inconnue du métal initialement présente dans l’échantillon (intersection de la droite d’étalonnage avec l’axe des abscisses) .
Ion pilote (différent)
(4) 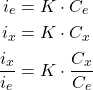
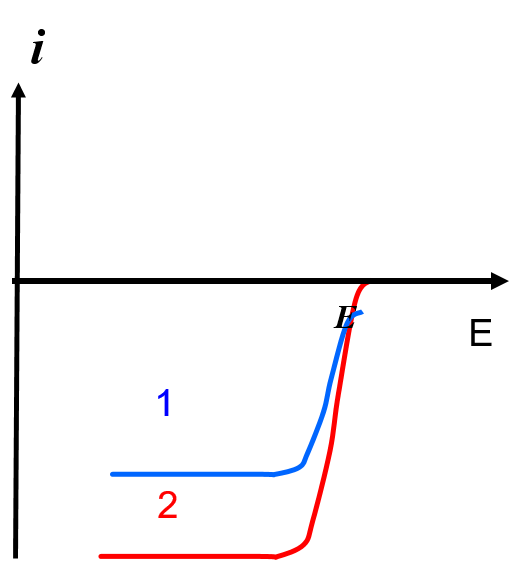
K ?
Faire un mélange d’étalons des 2 ions à la même concentration [c]
Mesurer les intensités. 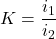
Application :
- Cations : Zn, Cd, Pb, Cu,…
- Organiques: Aldehyde, carbonyle, nitro …
- Médicaments: Antibiotiques; vitamines (B1, B2, B6) …
Exemple 1:
Exemple 2:
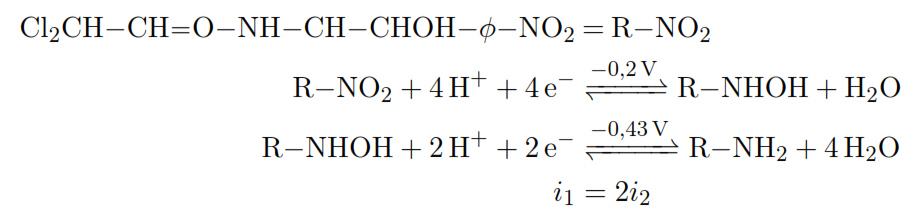
dosage de complexe métallique
i = f (MT) pour [L] fixe + [M] ↑
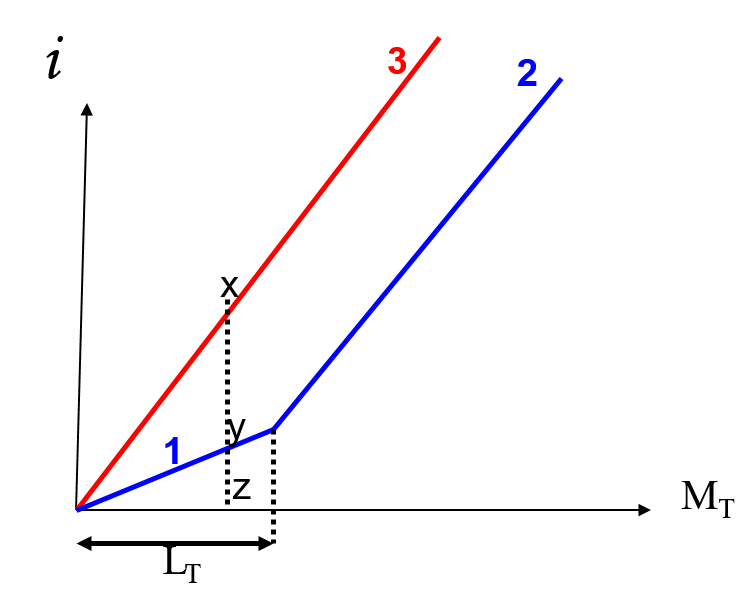
- (1): [M] quelconque, réponse de y par rapport à x (zone de Complexe)
- (2) M se comporte librement (saturation)
- (3) Évolution de Mt non interagissant avec L = (courbe d’étalonnage)
- LT: capacité Complexante de ligand
Détermination de Ks ( RUZIC)
(5) 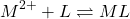
- (1) :
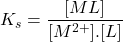
- (2) :
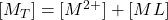
- (3) :
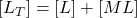
On démontre :
\begin{align}
\dfrac{[M^{2+}]}{[M_T] -[M^{2+}] } = \dfrac{[M^{2+}]}{[L_T]} + \dfrac{1}{Ks \cdot [L_T] }
\end{align}

Share this content:
Table des matières