Chromatographie en phase gazeuse (CPG)
Principe
La chromatographie en phase gazeuse (CPG) s’adresse à la séparation des constituants volatils ou volatilisables. Le mélange gazeux ou liquide est introduit dans le système d’injection, volatilisé s’il y a lieu puis entraîné par un gaz vecteur (phase mobile).
Selon leur affinité par rapport à la phase stationnaire, les constituants vont se séparer les uns des autres le long de la colonne, suivant une suite continue d’équilibres s’établissant entre le soluté et la phase stationnaire (liquide ou solide placée à l’intérieur de la colonne).
La colonne est placée dans un four thermostaté. C’est un tube de faible section, enroulé sur lui-même et contenant la phase stationnaire. Au bout de la colonne est placé un détecteur qui permet l’analyse sélective et parfois l’identification des mélanges.
Appareillage en CPG
Un appareil de CPG comprend schématiquement 3 modules spécifiques : un gaz vecteur, un injecteur(1), une colonne contenue dans une enceinte thermostatée: four(2) et un détecteur (3) relié à un intégrateur ou un ordinateur (4) sur lequel apparaît le chromatogramme .
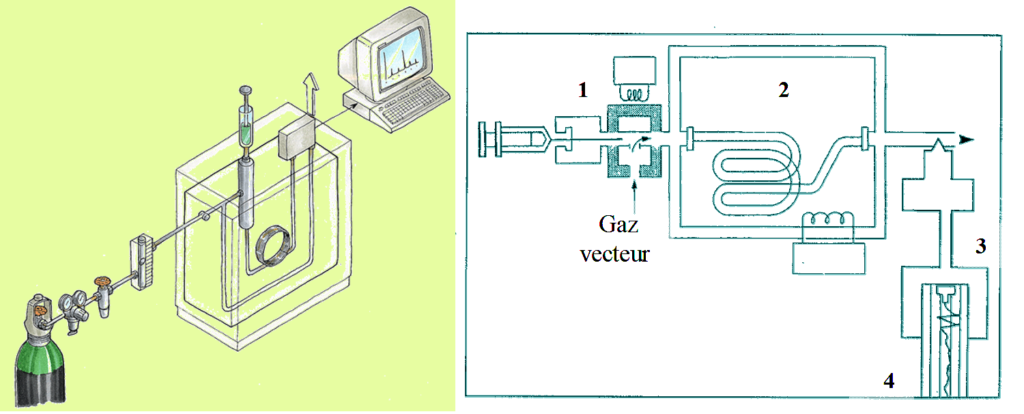
Le mélange à analyser est injecté sous forme d’un fluide, puis vaporisé dans l’injecteur. Le gaz vecteur l’entraîne dans la colonne de séparation thermostatée.
Les composés se répartissent différemment dans les 2 phases, et se déplacent donc à des vitesses différentes selon leurs affinités puis sortent à des temps différents. À leur sortie, ils sont détectés par le détecteur, et un pic apparaît sur l’enregistreur.
Gaz vecteur (réservoir)
Le gaz est contenu dans des bouteilles munies de manomètres.
Il circule dans la colonne à débit constant et il doit répondre à certaines qualités :
- Faible viscosité
- Grande pureté
- Inertie vis à vis de l’échantillon et de la phase stationnaire
- Compatibilité avec le détecteur
Les principaux gaz utilisés : l’azote, l’argon, l’hélium et l’hydrogène.
Système d’injection
Il permet:
- l’introduction, par le biais d’une seringue(1-10
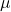 L), de l’échantillon,
L), de l’échantillon, - son évaporation (T° de l’injecteur = T° du produit le moins volatil + 20°C )
- son entraînement par le gaz vecteur vers la colonne
Injecteur à vaporisation directe
Pour les colonnes remplies et les colonnes capillaires de gros diamètre. Tout l’échantillon introduit par la seringue est entièrement entraîné dans la colonne. Risque de saturation des colonnes capillaires à faible débit.
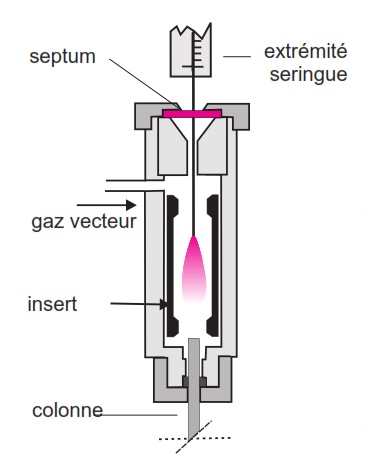
Injecteur avec système de fuite
Une grande partie de l’échantillon injecté, vaporisé et mélangé au gaz vecteur est éliminée de l’injecteur par une vanne de fuite. Ainsi, une petite fraction du mélange pénètre dans la colonne. Il existe deux modes selon que l’on injecte:
- vanne de fuite ouverte (mode split)
- vanne fermée pendant environ 1 minute après l’injection (mode splitless)
Injecteur à température programmable (PTV)
Le mélange est introduit sous forme liquide dans l’injecteur à froid puis l’injecteur est chauffé en mode split ou splitless pour vaporiser les composés.
Injection à froid dans la colonne
le mélange est injecté directement froid et liquide dans la colonne
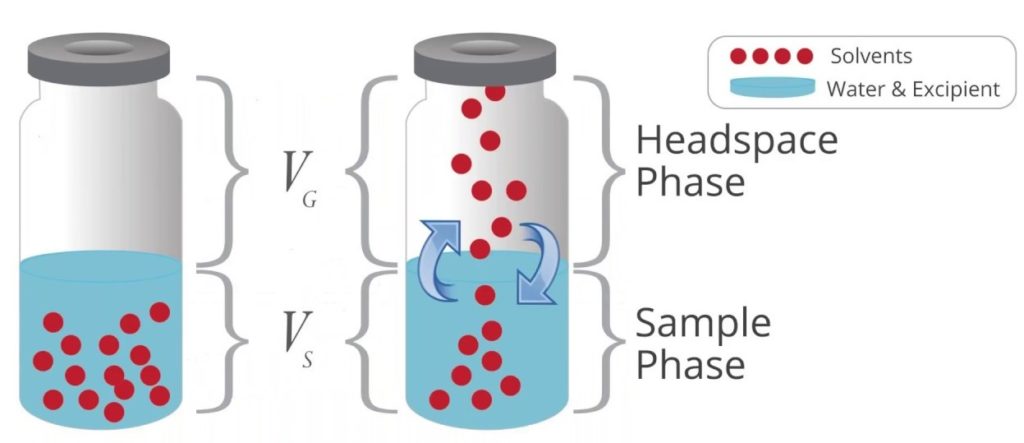
Technique d‘injection dite « espace de tête » ou « headspace »
Utilisée pour la détermination des constituants volatils dans des solides ou des liquides visqueux, échantillons non injectables. L’échantillon est chauffé dans un flacon fermé: les constituants volatils sont libérés et seuls sont analysés, sans influence des composés non vaporisables.
Four
Les colonnes sont placées dans des enceintes chauffées appelées four dont la température peut-être réglée au 1/10ème de °C près.
La température du four peut-être :
- Stable et identique du début à la fin de la manipulation (= conditions isothermes)
- Programmée par palier successif (mode gradient)
Colonnes
Elles contiennent la phase stationnaire, et se présentent sous forme de tubes fins enroulés.
Il existe deux types de colonnes :
Les colonnes remplies
Chromosorb® ; Sphérosil® ; Porapak®
Diamètre de 2 à 6 mm et longueur de 1 à 3 m. Elles sont en tubes d’acier ou verre.
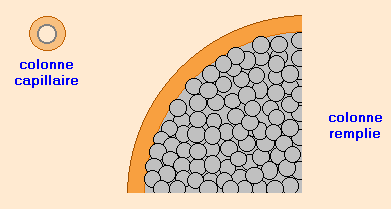
Elles sont remplies d’un support poreux et inerte sous forme de grains sphériques ( d’environ 0,2 mm de diamètre) sur lequel est imprégnée la phase stationnaire.
Moins résolutives que les colonnes capillaires.
Les colonnes capillaires (à tube ouvert)
Diamètre de 0,1 à 0,53 mm et longueur de 10 à 100 m. Elles sont en tube d’acier inoxydable ou en silice fondue ;
La phase stationnaire est directement déposée sur la paroi interne de la colonne sur une épaisseur de 0,05 à 5 m.
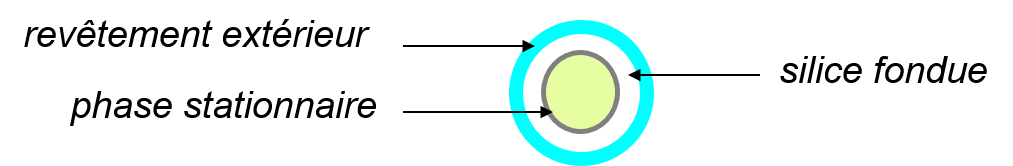
On distingue les colonnes:
- WCOT (Wall Coated Open Tubular) : la phase stationnaire = une pellicule liquide à l’intérieur du tube.
- PLOT (Porous Layer Open Tubular) : la phase stationnaire = une couche d’adsorbant solide.
Phase stationnaire
Choix de la phase stationnaire
- Une phase apolaire retiendra d’autant plus un composé qu’il sera apolaire (et inversement), Exemple : Squalane (apolaire) ; Carbowax (polaire)
- Une phase apolaire retiendra les composés dans l’ordre de leur température d’ébullition (donc sortie des composés dans l’ordre de leur température d’ébullition croissante)
- Une phase phénylée retiendra mieux un composé aromatique
Différentes phases stationnaires
Les phases les plus courantes sont:
Les polyéthylèneglycols (PEG)
polymères polaires (composés des colonnes Carbowax®) de type:
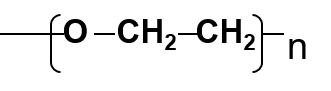
Les polysiloxanes (« huiles et gommes de silicones « )
correspondant à la répétition d’un motif de base de type:
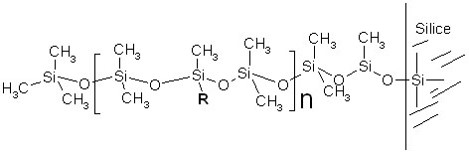
Suivant le pourcenage de groupement R par rapport aux groupes CH3, on peut modifier la polarité de la colonne et donc ses propriétés en chromatographie.
- Si R= CH3, la colonne est complétement apolaire et sépare les produits suivant leur point d’ébullition (noms commerciaux : DB-1, OV101, SE-30…)
- Si le pourcentage de R= Phényle est égal à 5%, on a la colonne la plus utilisée en CPG, elle est répertoriée sous les noms commerciaux suivants: DB5, CPsil5, OV5…
- Si on incorpore un substituant cyanopropyle (R= -CH2-CH2-CH2-CN), la polarité augmente beaucoup (à cause du fort moment dipolaire du groupe -CN). Ce sont les phases DB 1701, CPSil 18…
Ces phases à base de silicone présentent deux avantages pour la CPG :
- une bonne inertie chimique, elles ne réagissent ni avec les phases mobiles, ni avec les produits injectés.
- une très bonne tenue à la température, elles peuvent être chauffées sans dommage jusqu’à 300°C.
Détecteurs
Placés à l’extrémité des colonnes, Ils peuvent être plus ou moins spécifiques des composés à détecter.
T° du détecteur = T° du produit le moins volatil + 20°C
On détermine :
- Sa quantité minimale détectable (QMD)
- Sa sensibilité : correspond au rapport entre signal / concentration
- Sa linéarité
Il existe différents types de détecteurs comme le montre le tableau ci-dessous:
| Détecteur | Gaz vecteur | Linéarité | QMD | Sélectivité (spécificité) | Applications |
| Catharomètre | H2/He | 105 | 1 à 10 ng | NS | Tous composés |
| FID | H2/He | 107 | 20 à 100 pg | NS | Composés organiques |
| Capture d’électrons | N2 | 104 | 0,1 pg | S | Composés halogénés |
| Thermoionique | N2 | 104 | P :1 pg N :10 pg | S | Composés avec N ou P |
| Photométrie de flamme | N2/H2 | 103 | P : 10 pg S :1 ng | S | Composés avec S ou P |
Détecteur à ionisation de flamme (FID)
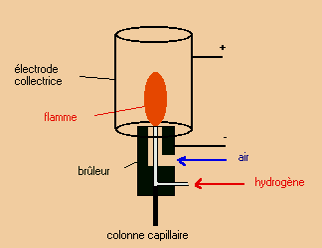
Le plus utilisé pour les composés organiques et de grande sensibilité.
Les composés (gazeux) qui sortent de la colonne pénètrent dans la flamme du détecteur. Leur combustion entraîne la formation d’ions et de particules chargées qui sont alors collectés par 2 électrodes. Le courant qui en résulte est proportionnel à la concentration.
Détecteur à conductibilité thermique (TCD)
Détecteur universel mais de sensibilité moyenne;
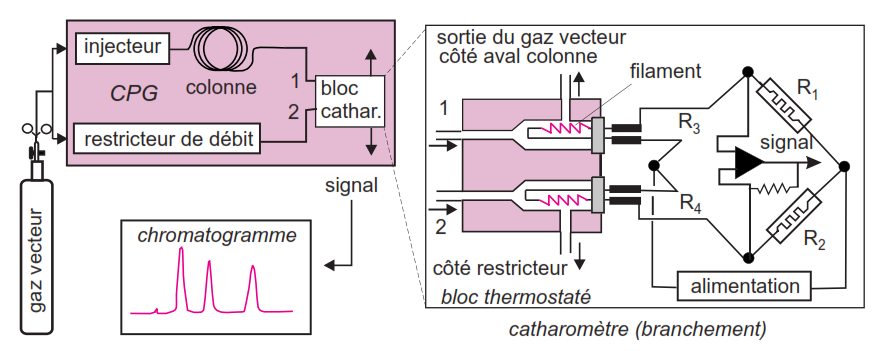
Il est formé d’un catharomètre composé de 2 thermistors (= filaments chauffés) dont un est balayé par le gaz vecteur seul et l’autre par le gaz en sortie de colonne; Quand un courant gazeux passe sur les filaments, ils sont refroidis en fonction de la température, du débit et de la nature du gaz; la température et le débit sont les mêmes pour le gaz arrivant sur les 2 filaments mais la présence d’1 composé en sortie de colonne modifie la nature du gaz qui alors refroidit moins le 2ème filament. Il en résulte une variation de résistance proportionnelle à la concentration du composé.
Détecteur thermoionique (NPD)
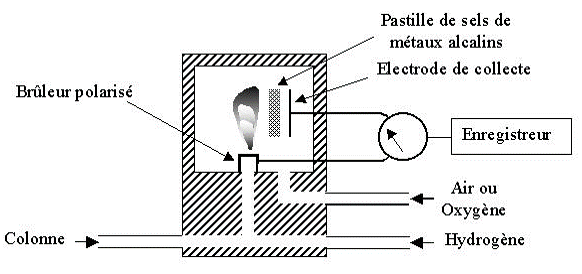
Un petit cylindre en céramique est chauffée par une résistance électrique à 800°C et est alimenté par un mélange air/hydrogène.
Les composés contenant N et P sont décomposés en ions négatifs qui sont recueillis par une électrode collectrice reliée à un électromètre qui traduit le courant obtenu en un signal.
Détecteur à capture d’électrons (ECD)
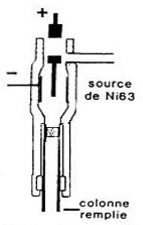
Spécifique des dérivés halogénés et très sensible.
Une source radioactive émet des particules  génère des électrons au contact d’un courant d’azote.
génère des électrons au contact d’un courant d’azote.
Ces électrons produisent un courant constant qui diminue lors du passage de composés contenant un halogène (F, Cl, Br) car ces composés captent une partie des électrons; la diminution du courant se traduit par un pic.
Détecteur à photométrie de flamme (FPD)
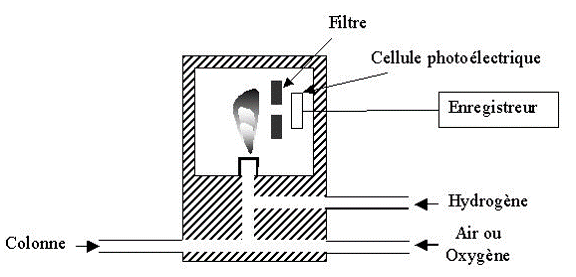
Les solutés élués sont brûlés dans une flamme très chaude air-hydrogène et les atomes de phosphores et de soufre sont excités : ils émettent alors une lumière verte ou bleue, détectée par un photomultiplicateur. Les radiations caractéristiques de ces 2 éléments sont sélectionnées par des filtres interférentiels ( = 390 nm pour le soufre).
Mise en oeuvre
Cas de composés difficilement séparables
Dans le cas de composés difficilement séparables, il est possible de recourir à une dérivatisation. Les réactions chimiques les plus couramment utilisées sont de 3 types :
Silyllation
Transformation en dérivé silylé, elle consiste à remplacer un hydrogène mobile par un groupe triméthylsilyle. La substitution permet:
- d’éviter la formation de liaison hydrogène , et l’association des molécules
- de diminuer la polarité des molécules,
- d’augmenter la volatilité des molécules.
Elle s’applique aux composés hydroxylés (OH), aminés (NH2) et carboxylés (COOH).
Les réactifs les plus utilisés sont triméthylchlorosilane TMCS: (CH3)3-Si Cl et hexamethyl disilazane
Alcoyalation
Cette réaction permet de remplace un hydrogène mobile par un radical alcoyle.
Elle transforme des alcools en éthers et les acides en esters.
Parmi ces réactions, on peut citer la méthylation.
Les réactifs les plus utilisés sont le sulfate diméthyle (CH3)2-SO4, le diazométhane CH2 = N+ = NH et l’iodure de méthyle CH3-I.
Acylation
Elle correspond à la réaction d’un cation acylium R-{+}C=O, sur les fonctions alcools ou amines donnant naissance à des esters ou des amides.
Les dérivés obtenus sont très volatils notamment lorsqu’ils renferment des halogènes comme les dérivés perfluorés.
Les réactifs les plus utilisés sont : chlorure d’acide et les anhydrides d’acides.
Cas de composés facilement séparables
Dans le cas de composés facilement séparables, l’échantillon prêt à l’emploi est injecter directement sans dérivatisation.
Applications
- Biologie clinique: Dosage de cholestérol, détection des acides gras .
- Pharmacologie: Dosage des benzodiazépines, des métabolites médicamenteux pour des études de bio-disponibilité et de suivi thérapeutique.
- Toxicologie: Méthode officielle de dosage de l’éthanol. Dosage du méthanol, éthylène, glycol, cyanure, les carbamates.
- Pharmacie hospitalière: Dosage de l’oxyde d’éthylène dans le matériel médico-chirurgical stérilisé par ce gaz. Contrôle des gaz médicaux , matières premières, produits finis et les alcaloïdes.
- Industrie agroalimentaire: Dosage des pesticides, recherche des nitrosamines dans l’atmosphère et dans les produits de charcuteries.
Analyses chromatographiques
Analyse qualitative
- L’identification est assurée par la mesure de tr
- La confirmation : par couplage CPG – SM.
Analyse quantitative
Basée sur la relation:
\begin{align} C_i = K_i\cdot A_i \end{align}
Ci : Masse de la substance i ayant traversé le détecteur
Ai : Aire de pic correspondant
Ki : Facteur de proportionnalité ou coefficient de réponse
On distingue trois méthodes principales :
- Normalisation interne : On compare les aires des pics d’élution au cours d’une même injection.
- L’étalonnage externe ou « méthode des injections comparées « : On compare sur deux chromatogrammes l’aire du pic d’élution de la substance de référence à celle du produit à doser.
- L’étalonnage interne : On compare les aires des pics de la substance de référence et du produit à doser à la surface du pic d’élution d’un produit témoin appelé « étalon interne » ajouté lors du dosage et de l’étalonnage.
- Étalonnage par ajouts dosés : On ajoute à une concentration inconnue mais constante, de notre composé des ajouts successifs de substance à analyser.
Normalisation interne
Tous les pics sont élués :
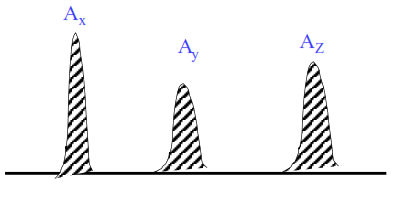
\begin{align}\% X =\dfrac{f_x \cdot A_x }{f_x\cdot A_x + f_y\cdot A_y+ f_z\cdot A_z}\end{align}
idem pour y et z.
f : facteurs de réponse déterminés par rapport à un composé de référence fR = 1,000.
Étalonnage externe
Cette méthode est basée sur la comparaison de deux chromatogrammes (étalonnage et dosage) effectués dans des conditions identiques.
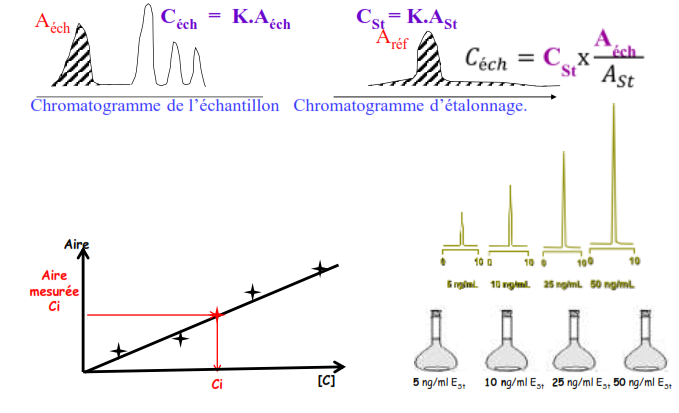
Problèmes:
- Il peut y avoir des problèmes de répétabilité du volume injecté
- la référence est indispensable et peut être indisponible dans le commerce
- Effets de matrice
Etalon interne
On rajoute au mélange une quantité connue d’un corps pur (EI), celui-ci doit avoir:
- Structure chimique très proche
- Temps de rétention très proche
- Concentration très proche
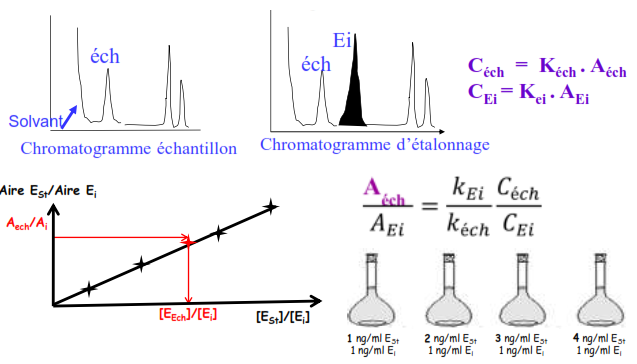
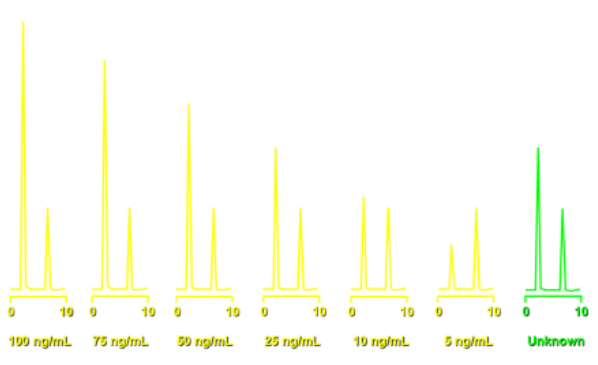
Étalonnage par ajouts dosés
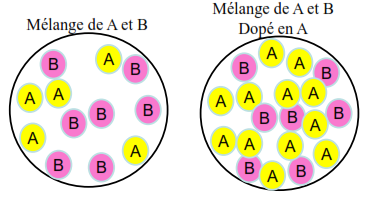
On veut doser A:
- Le détecteur doit détecter A Mais B interfère car le détecteur détecte un peu B, Alors on aura un signal correspondant à la quantité de A et un peu de la quantité de B, Du coup, [A] sera surestimée
 (Effet de matrice)
(Effet de matrice) - L’échantillon est dopé en A (concentration connue) B interfère toujours, Mais l’effet de B est réduit jusqu’à négligeable. Du coup, [A] n’est plus surestimée. (Suppression de l’effet de matrice)
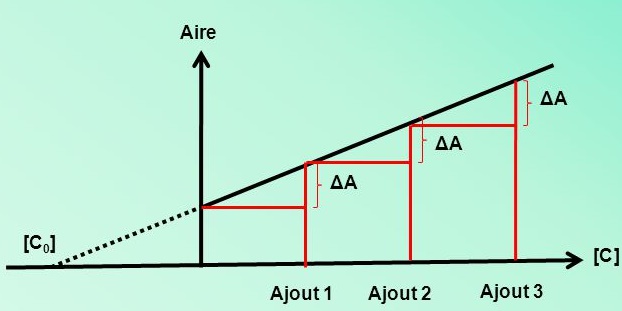
Ajouter à une concentration inconnue mais constante, de notre composé des ajouts successifs de substance à analyser.
Aire. [C ] [C0] Ajout 1. Ajout 2. Ajout 3. ΔA.
Share this content:
Table des matières

